

有水有渝
第1楼2009/12/15
下面举一个经过酯化、胺化和水解三步反应的跟踪检测:
液相色谱仪:岛津LC-10A或LC-20A液相色谱仪,LCsolution或浙大N2000工作站。
色谱条件:岛津C18(250mm*4.6mm,5um),以30mmol/L磷酸二氢钾溶液(用磷酸调PH=3.0)-甲醇(70:30),检测波长:215nm,流速1.0ml/min
反应液组成:原料(含有酚羟基和侧链醇羟基,为可酯化部位),酯化基团(甲磺酰基),溶剂(乙酸乙酯)等。
A、酯化反应液
反应20h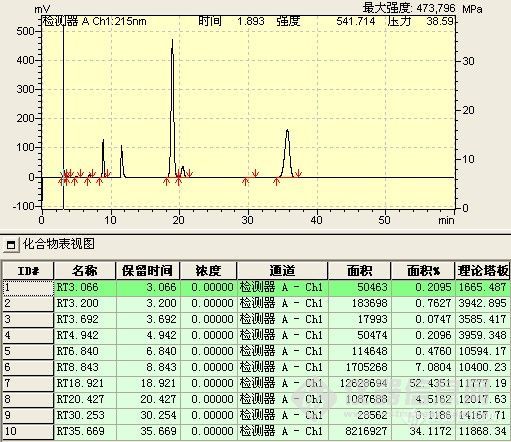
其中,8.843min为原料,18.9min与35.6min均为所要产物,前者为醇酯化物,后者为酚羟基和醇羟基均被酯化,20.4min为酚酯化物(副产物),10-20min之间有一未积分色谱峰为溶剂乙酸乙酯峰。
反应28h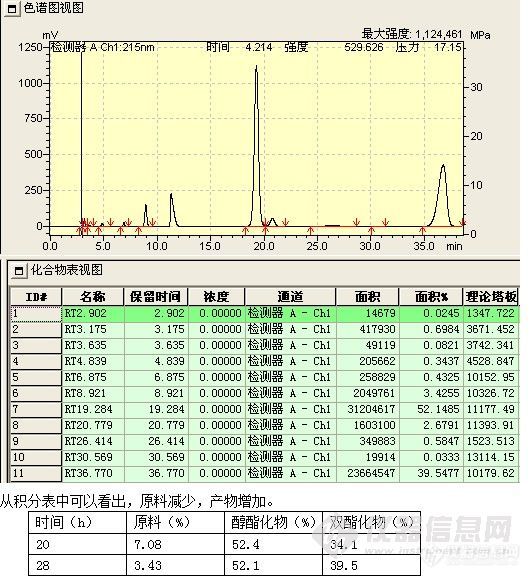
原料的减少量约等于产物的增加量,随着时间延长,单酯化物有向双酯化物转化的趋势,但副反应并没有明显增加。
醇酯化物波长扫描,把上图的峰高与吸光度对照基本一致。
2、 组分的变化 主要观察除主成分峰外其它色谱峰的变化,可以很好反应副反应的变化,当然不排除反应过程中有中间态的产生,要加以区分,这也是选择反应条件的重要因素之一,如温度,反应液中各组分的比例,反应时间等。有机合成首要的不是反应要多快的完成,而是减少副反应,利于提纯,只要能够原料反应完全,副反应少,那么反应条件弱一点,反应时间长一点,都是可以接受的。对于我们检测人员来说,要做好以下方面的记录:(1)各有关物质的保留时间,峰形,各组分所占的比例,以便于下次对它们作出判断,是否副反应增加,有无新的杂质产生。由于每次流动相配制,温度等因素影响,保留时间可能引起变化,可以主成分峰为基准,引入相对保留时间来确定各组分位置;(2)峰面积 可用归一法与以前的相同条件下的反应液比较,产物是否正常,副反应是否增加,是否有新的杂质产生,各峰面积的比例是否产生变化,有变化时,应及时反馈,与合成人员进行分析。(3)这里要说到一点,更换色谱柱时,组分的出峰时间可能有变化(在一次分析喹啉类化合物时,其原料和产物在奥泰C18柱与岛津C18柱上的出峰顺序正好相反)这一点要加以注意,发现与以前不同的,也不要惊讶,重复进一次看看,必要对各组分重新进行定性。(4)注意可能会出现反应向负方向进行的情况,在分析某一喹啉类化合的反应,其步骤为原料经付克反应生成产物1,产物1再胺化生成产物2,结果检测胺化反应液时,发现与原料相同保留时间处峰面积明显增强(初始仅能判断可能是原料,有待确定),经过分析,确定为付克反应没有得到正确的产物,喹啉环上有一羟基,所要产物付克位置应为羟基的对位,但结果却是先与羟基进行了反应,在胺化碱性条件下,水解回原料,最后得出,应增强付克反应条件和反应时间(利于付克基团的重排)。
有水有渝
第2楼2009/12/15
B、酯化反应催化剂不同,引起反应选择性不同,下图为在另一催化剂下组分变化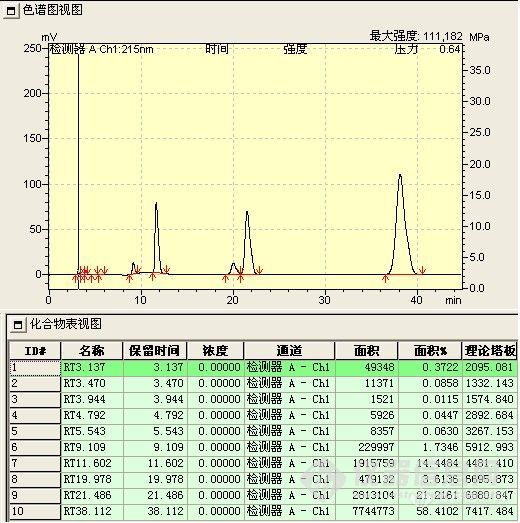
酚酯化物(副产物)明显增多。
C、胺化反应液 按理论,在反相色谱上,化合物胺化后,其极性应增强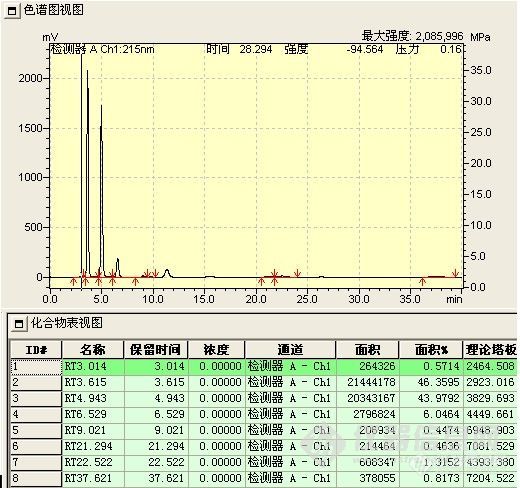
理论与实际符合,3.6min为醇酯化物的胺化产物;4.9min为双酯化物的胺化物产物,该化合物水解酚酯端成酚羟基,转化成3.6min的胺化物(最终产物)。
D、酯化产物比例不同,引起水解反应完全的难易程度不同。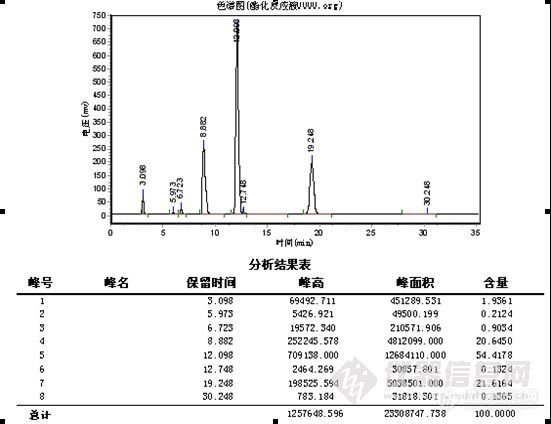

上图 保留时间提前是由于为缩短分析时间而增加了甲醇的比例至38%后的结果醇酯化物:双酯化物=2.5:1
下图 醇酯化物:双酯化物≈1:1,大大增加胺化后水解的时间,且水解难以反应完全。反馈的结果是应促进醇酯化物的生成,抑制双酯化物的生成,以此方向来优化反应条件。
E、水解反应液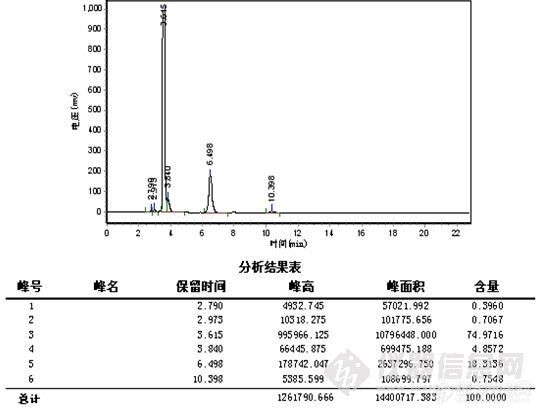
水解完全,在5min左右未见明显双酯化的胺化物,6.5min为副反应产物。
有水有渝
第3楼2009/12/15
F、反应往负方向进行的例子
a、付克反应液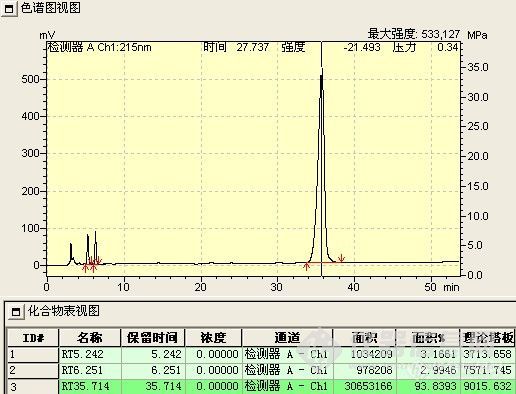
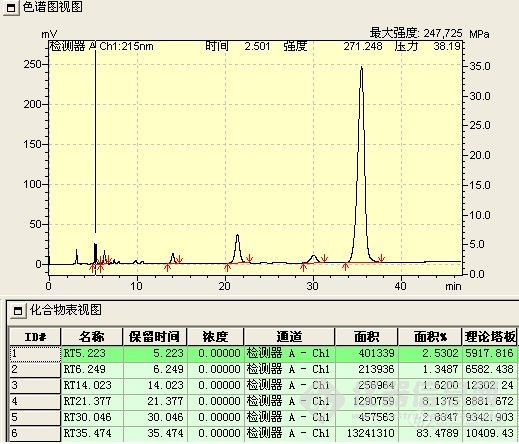
上下两图仅反应时间不同,下图反应时间长,其中5.223min为原料,21.377min处为所要产物,35.474min处为中间态(开始时被误认为是所要产物)
b、胺化反应液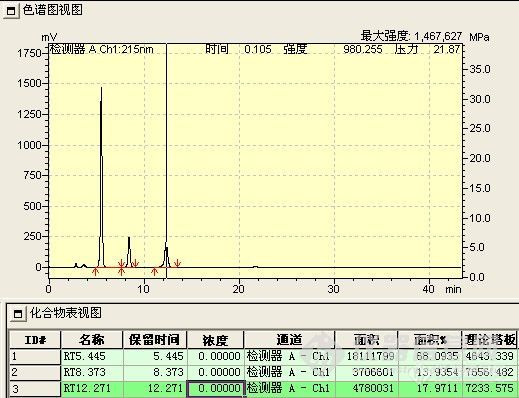
胺化后,原料峰明显增强,中间态峰消失。反应实际结果与理论不符。
3、保留时间变化 保留时间变化要分析是组分漂移还是有新的组分产生,如果是环境对保留时间时间影响比较大的,也可将其与需要对比的反应液混合以确定是同一个物质还是其它新的物质。
三、 成品阶段
此阶段主要是对提高产品纯度的相关处理,主要方法有萃取,蒸馏,重结晶,过柱等。这一阶段有归一法纯度检测和对照品法含量检测两种情况。
1、 纯度与含量基本一致 这种情况对于我们跟踪分析人员来说是乐于见到的,如果多次结果均一致,则跟踪时以归一法来检测就行,省时,省力。
2、 纯度与含量不一致 这种情况基本上是纯度高于含量(常以主成分最大吸收为检测波长)。出现这种现在的可能是:(1)检测波长选择不当,主成分峰的响应强于有关物质的响应,可用自身对照法或引入校正因子的方法来解决,规定校正因子在0.9-1.1范围内的,可用直接归一法;若选择的波长是有关物质响应大于主成分,则可能出现纯度低于含量的现象。(2)测纯度时,进样浓度太低,有些杂质浓度低于检测限而未被检测出,增加进样量可以解决。(3)成品中可能含量不响应的盐类杂质,有机溶剂或水分等。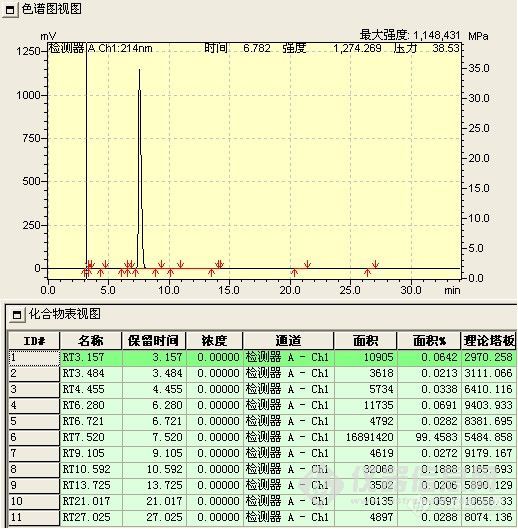
某化合物归一法含量为99.5%,但按标准以内标法检测,实际含量为98.2%,经与合成人员分析,可能是其中的铵盐没有除尽。(色谱条件:岛津C18柱,50mmol/L磷酸二氢钾溶液(用磷酸调PH为3.0)-甲醇-乙腈(55:34:11),检测波长214nm,柱温30℃)
四、 结束语
以上是本人的一些分析总结,有不足之处,欢迎版油批评指正,不断完善本人的分析方法。
xianer1997
第9楼2009/12/18

































