-
+关注
私聊
-

皮皮鱼
第11楼2009/04/13
呵呵,你说的不完全吧,是不是有保护反切啊,那个反峰是不是切阀扰动哦。
你的谱图非常好,没有你说的峰型不对称的问题,很对称。甲烷很难不对称的。
你的问题是基线发生切阀扰动,甲烷峰出在扰动上,导致看上去峰型畸变了。等等,你的基线这么上山下乡的?为什么啊?
明白了,你没有切阀保护,你的柱子就是一根分子筛。前面的反峰是你样品中的高纯N2,基线的上山下乡是你样品中含有的重烃。你是分析高纯N2中的微量烃类吧?你的标气或者样品中含有重烃吧?由于你直接分析,这些重烃在你的色谱柱中存留,流出速度很慢,形成了你的基线长周期扰动。估计这些重烃来自你的标准气。
可惜你无法得到稳定的基线,否则你背景扣除之后,你会发现你的甲烷峰非常良好的。峰型这么难看,是你的基线难看,不是峰难看啊。
解决办法:老化色谱柱过夜,第二天来了空走基线,看看是不是直了。如果直了,马上分析样品,这一针可以保证峰型是好的了。后面的就不好说了,如果你这一针里面有重组分进去了,后面基线就已经乱了。呵呵。
怎么解决?没法解决。除非你更换方法,保护一下柱子,让重烃进不去,或者进去了反吹出来。例如前面用Porapak-Q柱子保护一下,或者用下面的气路反吹重烃出柱子,才成。
这个是做非甲烷总烃的方法,甲烷出峰立刻切阀反吹,2倍甲烷出峰时间后出其他烃类的一个合峰。合峰出完全了就可以分析下一个样品了。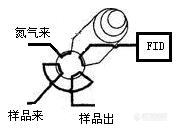 xiana1978 发表:其他分析条件:柱子为2m的5A分子筛填充柱,柱温为80度, FID的检测器温度为200度,载气为高纯氮气,总流量为30ml/min,氢气流量为40ml/min,空气流量为400ml/min,背景气是大气.进样方式为六通阀进样.
xiana1978 发表:其他分析条件:柱子为2m的5A分子筛填充柱,柱温为80度, FID的检测器温度为200度,载气为高纯氮气,总流量为30ml/min,氢气流量为40ml/min,空气流量为400ml/min,背景气是大气.进样方式为六通阀进样.
-
+关注
私聊
-
皮皮鱼
第12楼2009/04/13
恩。你这三个图是在连续分析,后面看到的反峰是下一次分析的反峰了。空走之所以没有反峰,是因为后面还没进样。
你的载气可能也有问题,为什么样品中高纯N2出反峰呢?六通阀进样绝对不会有这个的啊,看来是载气纯度差了。你是不是用的老CO、CO2分析的仪器改造的?甲烷化器没去掉?这个反峰挺有趣,据我分析是载气中含有出峰在甲烷之前的非甲烷烃类才能这样。因为这个反峰在甲烷峰前面啊,这么一看就只有CO了,还要有甲烷化器才能出现这个样子呢。否则哪里有出峰在甲烷前面的烃类。恩,继续分析,那么你这个甲烷化器现在不受保护了,重组分可以进去。甲烷化器进了重组分,特别是温度又没有足够高(400度),比如在300度左右,那么就出现这个基线大范围拨动了。同时,这时候也能对CO进行有效的转化,确保进样能出反峰。如果你的重组分是乙烯或以上,那么400度也会基线波动。
恩恩,就像在猜谜语哦,好玩。如果我猜中了,那么把甲烷化器拆了吧。拆了以后,重组分出峰快的多,可能分析周期长点就没有波动了。
哦。还有,通过适当的数据处理,例如积分参数调整,应该可以消除这个干扰的。但是你第三张谱图太吓人,不太好设置这些参数了。
-
+关注
私聊
-
皮皮鱼
第13楼2009/04/13
再看看还是觉得不对。反峰可以解释为载气不纯,第1和第2张图中后面的基线骤降怎么解释?还是有切阀保护的啊,这个只能用切阀扰动来解释啊,难道你还做了灵敏度调整或者基线强制?那么说就是你保护晚了,切阀动作慢了,才让重组分进入甲烷化器的。嘿嘿,如果我又猜对了,那么调整这个切阀时间可以保护好呢。
FID,载气和样品本底不同,比如分别是N2和He,虽然对离子化效率有影响,但不会出这么大反峰的。
胡乱猜测,楼主看到了谈谈实际情况哦。xiana1978 发表:其他分析条件:柱子为2m的5A分子筛填充柱,柱温为80度, FID的检测器温度为200度,载气为高纯氮气,总流量为30ml/min,氢气流量为40ml/min,空气流量为400ml/min,背景气是大气.进样方式为六通阀进样.
-
+关注
私聊
-
皮皮鱼
第19楼2009/04/13
他基线波动相当不小啊。就他给的基线,波动范围在0.4了,而他的色谱峰才0.65高哦。
仔细看他的谱图很久,似乎发现了昨天晚上没看到的东西,昨天看来确实太晚太困了。
它的基线陡降其实是色谱分析开始,内部程序做了一个基线强制回零。这说明他确实就是六通阀,没有保护反吹措施的。
前面的朋友为反峰给出了很多理由,还是有点不理解。柱前压115kPa,1ml定量环会造成基线这么大的下降?空气湿度会造成固定每次分析都有下降,而空走基线就没有?
古怪。这几张谱图我收藏了。很有趣,很有分析的价值。
当然,他的问题答案很明白:
这些谱图中甲烷峰其实很好很对称,只是基线波动太大了,导致甲烷峰看上去很难看。orangeghost 发表:呵呵,不好意思,后来图片打开时间窗看到了,其实你的空白应该还算错的,因为你尺度放的大,所以看着就比较起伏