-
+关注
私聊
-

yhl-87_
第103楼2009/11/03
顶空气相色谱系列讲座(99)
在线分析脱丙烷塔顶中气体组分的气相色谱柱系统研究
摘要: 介绍一篇解决生产中工艺气体的在线分析方法。根据生产要求,对裂解装置中高压和低压脱丙烷塔顶中工艺气的气相色谱在线监控柱切换流程进行了研究。通过3个进样器的二次异步进样,结合前吹、反吹等技术,应用7根柱子,成功地在6min内实现了对C2S、丙烷、丙烯、丙炔、丙二烯及C2S的在线分离分析。现场应用表明,研制的柱切换系统满足工艺监控要求。
1、 前言
气相色谱由于具有快速、简单、高效、高分辨率等优点,已被石油、化工、轻工、环保等领域的分析实验室广泛使用,并在许多现代化的大生产中,作为过程控制仪器日益显示出重要性⑴~⑸。根据我们的生产要求,拟对裂解装置中的高压和低压脱丙烷塔顶气进行在线色谱监控,在有关研究结果⑹~⑻的指导下,我们成功地建立了柱系统和切割时间程序。实践表明,研制的柱系统满足生产控制需要。
本文给出的这些研究成果是由广州石油化工总厂乙烯分厂的蔡华、刘漓江和茂名石化乙烯工业公司的严健以及中国科学院大连化学物理研究所国家色谱研究分析中心的路鑫、叶芬、许国旺等研究人员共同完成,现全文介绍如下:
2、实验部分
2-1 仪器和试剂
本实验所用气相色谱仪为上海科创色谱仪器公司的GC-900和美国ABB公司的过程分析仪,后者带3个进样器、1个氢火焰离子化检测器(FID)和1个热导检测器(TCD)。固定相从美国ALLTECH公司购得。所用柱管均为不锈钢管。色谱柱按国家色谱中心标准生产。
2-2 待监控的工艺气的基本组成
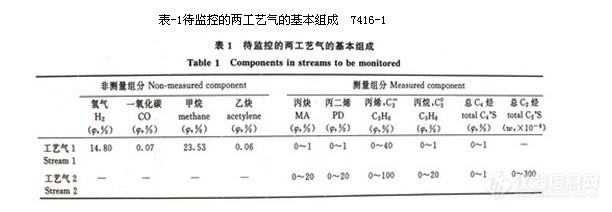
根据要求,本研究要监控裂解装置中高压和低压脱丙烷塔顶工艺气中的组成。基于掌握的技术资料,两工艺气的主要成分为H2,CO,C1,C2,C3,C4和少量的重组分,其中C2为痕量(0~300×10-6),其余组分为常量(见表-1)。要求监控的组分为C2,C3和C4,且要详细知道丙烷、丙烯、丙炔、丙二烯每个组分的含量,而C2和C4则只要测出总量即可。
3、结果与讨论
根据上述工艺要求及可采用的ABB在线分析仪的配置,拟采用如下的基本设计思想:
(1) 用高灵敏度的FID检测器测痕量C2;
(2) 充分利用3个进样口,以减少共存组分相互重叠而对柱系统选择性的过高要求。为此,首先将其分成两大子柱系统(简称子系统)来考虑。
子系统-1:它的主要任务是分离并检测工艺气2(表-1)中的痕量C2组分,需要将C2与共存的极微量轻组分分开并反吹C2后的重组分。我们选用了两根色谱柱来达到这样的分析目的。柱1为预分离柱,用来反吹C2后的重组分;柱2为主分析柱,用来实现C2和轻组分的分离。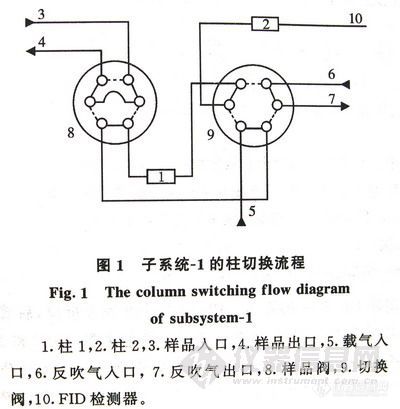
图-1给出了子系统-1的柱切换流程。7417-1
子系统-2:它的任务是分离两工艺气中常量的4个C2组分 (丙烷、丙烯、丙炔、丙二烯)和C4总烃。需前吹样品气中的轻组分H2,CO,C1和反吹C4后的少量重组分。由于4个C3组分要求全分离,难度较大,本设计方案中使用硅胶键合固定相(Phenyl isocycanate/Porasil C)作为分析柱,使4个C3组分得到了很好的分离。但C4组分在这个主分析柱上的保留时间过长,导致分析周期过长,不能满足工艺监控的需要。
在研究过程中发现,在上述柱子上丙二烯和丙炔之间的分离度很大。能否再增加一套可在短时间内分离出C4的子柱系统,分别用来分析4个C3组分和C4组分? 答案是肯定的。具体实施时可通过两次进样,调整两个子柱系统的进样时间,使C4组分的出峰时间穿插在丙二烯和丙炔之间,这样C3和C4组分均能得到很好的分离,并使分析周期大大缩短。根据这样的设计思想,我们选用了5根色谱柱来作为子系统-2的柱系统,并用TCD作检测器。柱切换流程见图-2。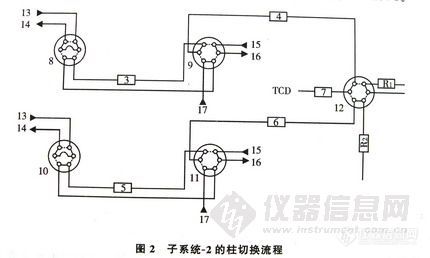
图2子系统-2的柱切换流程 7418-1
l气阻,2.R2气阻,3.柱3,4.柱4,5.柱5,6.柱6,7.柱7,8.样品阀,9.切换阀,10.样品阀,l1.切换阀,12.切换阀,13.样品入口,l4.样品出口,15.反吹气人口,16.反吹气出口,l7.载气入口。
其中柱-3和柱-5为预分离柱,柱-4和柱-6为主分析柱;柱-3、柱-4和柱-7为一套柱系统,用来分析C-3组分;柱-5、柱-6和柱-7为另一套柱系统,用来分析C4组分;柱7为两套柱系统共用,以进一步改善分离。在柱5上的进样时间是否合适是成败的关键。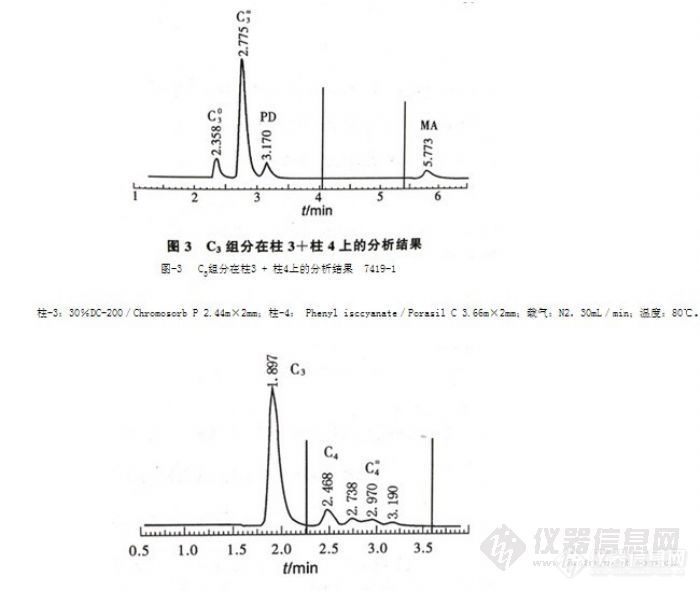
图-4 C3、C4组分在柱5+柱6上的分析结果 7420-1
柱5:30%DC-200/Chromosorb P 0.30m×2mm;柱6:30%DC-200/Chromosorb P 4.88m×2mm.;载气:N2,30mL/min;温度:8O℃。
图-3给出了C3烃在柱-3、柱-4上的分离谱图。图4则为相关样品在柱-5和柱-6上的谱图。从图-3,4中可知,PD和MA之间可供容纳C4的区间为4~5.5min (见图3),而C4烃在柱-5、柱-6上的保留时间为2.2~3.6min (见图4)。因此,在柱5上的进样时间应比在柱3上的进样时间拖后1.8min左右,即在柱5上的进样时间为85~130s。
用所研制的气相色谱柱系统分离丙烷塔顶中气体组分的色谱图见图-5,可以看出该柱系统与预先的设计思想相符,子系统-1中FID检测出的C2峰出在子系统-2中流出的C3峰前,而C4则正好插在PD和MA之间,总C4烃为iC04 + nC04 + C4=,结果较为理想。应该指出,由于在ABB在线分析仪中所用的切换阀为十通阀,因此,子系统1和2的实现只需4个十通阀。
经现场实验,保留时间的重复性在1%以内,峰面积的重复性为1%~3%,能满足工艺要求。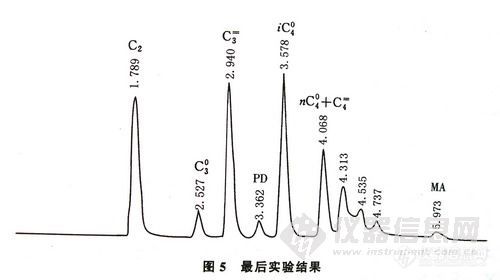
切换程序:柱1反吹时间70s,柱3反吹时间90s,柱5反吹时间157s。载气:H2 (柱I通道),N2 (柱-3、柱-5通道),30mL/min;温度80℃。
柱-1:30%DC-200/Chromosorb P 1.8m×2mm i.d.
柱-2:30%DC-200/Chromosorb P 3m×2mm i.d.。
柱3、柱4参见图3;
柱5、柱6参见图4;
柱-7:20%DC-200/Chromosorb P 1.2m×2mm i.d.
参考文献
1、 Hernan J Cortes.J Chromatogr,1992,626:3-23
2、 Bertsch W.HRC&CC,1978,1:85,187,289
3、 Jennings W.HRC&CC ,1979,2:441—443
4、 Huskins D J.Gas chromatographs as industrial process analysis.Bristol: Adam Hilger Ltd,1977
5、 De Geus H J,de Boer J,Brinkman U A Th.TRAC,1996,15:168~178
6、 卢佩章,许国旺.气相色谱专家系统.济南:山东科技出版社,1994
7、 许国旺,路鑫,汪敏燕等.色谱,1999,17(2):115~118
8、 许国旺,杨黎,刘辉等.色谱,1995,13(5):310~315
-
+关注
私聊
-
yhl-87_
第104楼2009/11/03
顶空气相色谱系列讲座(100)(上)
两种轻烃分析方法(“PTV切割反吹矫口“顶空”)的对比研究
摘要:本文由中国石油天然气股份公司油气地球化学重点实验室的肖廷荣, 蔡冰, 孟建华, 王培荣等高级工程师们建立的“程序升温蒸发进样器(PTY)切割反吹”和“顶空”两种有关原油中轻烃的分析方法,可延长色谱柱的使用寿命、分析周期缩短了,原油中<C9的较轻组分的分离度能达到美国材料试验学会(ASTM)D5l34-98所要求的标准,并有良好的重复性。此外,还报道了采自我国6个油区的l0个油样用两种分析方法所得19个地化参数的相对误差范围约为±(1%~25%),并对引起误差的原因进行了探讨。指出用这两种方法所获得的数据是不能合在一起作地化研究的,将原油直接注入汽化室所得的分析数据较可靠,顶空分析数据在用作地化研究时应加以校正的结论。
1 前言
近几年来,地球化学研究工作中有关轻组分研究的论文明显增加 [1],我国也己将轻组分化合物的研究纳入原油分类、油源对比等研究工作之中[2],有关人员己逐步认识到原油是由“较轻”(<nC9)、“中等”、“较重”(nC20)的碳氢化合物混合而成,大部分“生物标志物”是处于“较重”分子质量的范围,在高成熟原油或凝析油中其含量往往不到百分之一,甚至接近于零,而“较轻”分子质量的则可能占50%以上。显然,在研究“生物标志物”的同时应重视研究轻组分化合物,这样才能较好地掌握原油的“全貌”,这一点在研究混源油时尤为重要。
轻组分化合物一般采用色谱分析[3,4],我国所用的地化数据的分析方法有“全油色谱”、“油顶气”等。本文介绍的“程序升温蒸发进样器(PTV)切割反吹”(与“全油色谱”相似)和“顶空”(与“油顶气”分析法相似)两种分析方法,分辨率能达到国际水平[4],且色谱柱的使用寿命长,分析周期短。
2 实验
2.1 分析方法
2-1-1 PTV切割反吹分析法 仪器与色谱条件:
美国Agilent 6890气相色谱仪,配有两套电子压力控制器(EPC)及PTV装置(见图1)。Agilent生产的50m P0NA柱;载气为氦气;柱温为35℃(30min)以2℃/min升至60℃,再以8℃/min升至200℃(15 min)。汽化室温度为200℃(30 min),以50℃/min升至350℃(45min)。检测室温度为250℃。分流比为1:200,保持2 min后改为节约方式的分流比1:15。
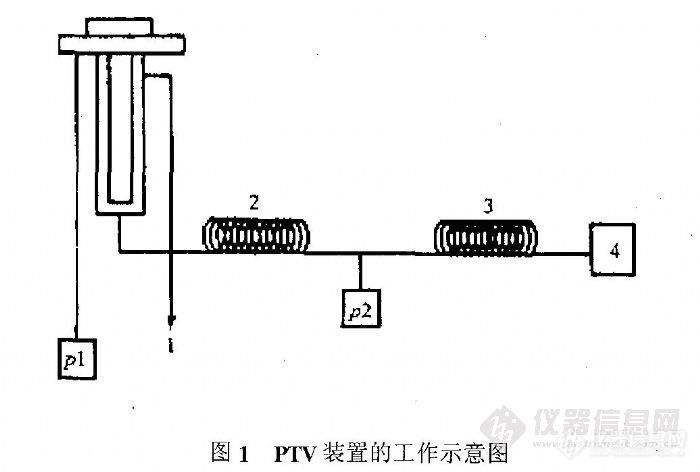
由图1所示的PTV装置的工作示意图可以看到,开始设PTV为200℃,压力为pl>p2,样品随载气流入色谱柱。当C8以前的组分全部流入分析柱,而较重组分基本上仍保留在前置柱时,进行反吹,即设PTV由200℃以50℃/min的速率升至350℃,压力改为p1
2-1-2 顶空分析法
仪器与色谱条件:意大利的FISONS 8500气相色谱仪,配有HS800顶空自动进样装置。色谱柱的型号及柱温同2-1-1项;汽化室温度为220℃;检测室为250℃;分流比为1∶120。样品加入密封的瓶中加热60℃,恒温10 min,取样品瓶中顶部的气体注入汽化室。
2.2 分离度
2-2-1 PTV切割反吹分析法
经过逐步试验,筛选出可使大多数色谱峰基本达到基线分离(见图2)的最佳色谱条件。在最终选定的2-1-1项所列色谱条件下,难分离的物质对2-甲基己烷和2,3-二甲基戊烷(见图2中的6-9峰与6-10峰)的分离度达到70%以上;一般很难分离的3-乙基戊烷和反-1,2-二甲基环戊烷也能显示出来(见图2中的6-15峰与6-16峰),其分离度达到ASTM D5l34-98所要求达到的标准[5]。
对图2中的各种轻组分采用Aldrich生产的正己烷、2,⒋-二甲基戊烷、2-甲基己烷、2,3-二甲基戊烷、3-甲基己烷、正庚烷、甲基环己烷、乙基环戊烷、甲苯等9种标样及文献[5]所列的Kovats指数对照定性,定性结果见表1。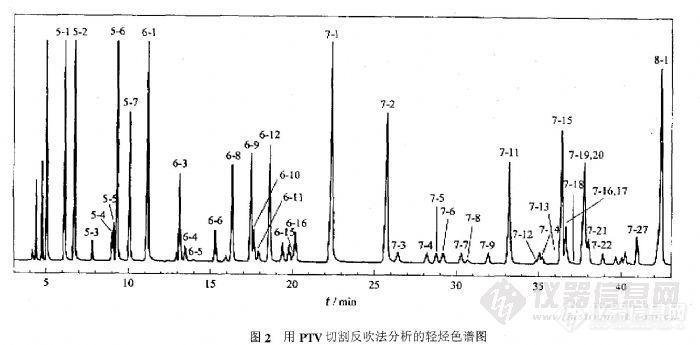
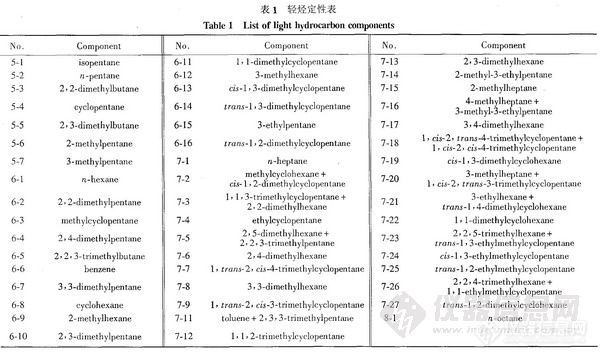
2-2-2 顶空分析法 采用2-1-2项所述色谱条件作油样的顶空分析,分离效果基本上达到文献[2]所要求的水平,仅3-乙基戊烷处分离效果略差,只出现了—个肩峰(见图3)。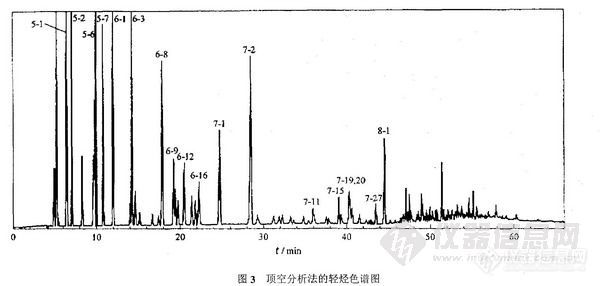
2.3 重复性
2-3-1 PTV切割反吹分析法
对上述9个标样的混合标准溶液进行3次平行测定,测得9个标样组分的质量分数范围为6.44%~14.48%,其相对偏差(8.5%,在SY/T 5120-1997规定的色谱分析误差范围之内[6]。
用塔里木轻质原油作重复性试验,峰编号为5-1至8-1共62个峰的3次分析结果的平均相对偏差分布范围为0.22%~8.53%,平均值为2.15%。
2-3-2 顶空分析法
对9个标样的混合标准溶液进行3次平行测定,测得9个标样的质量分数范围为6.44%~14.48%,平均相对偏差范围为2.03%~7.88%,均在SY/T 5120-1997规定的色谱分析误差范围之内[6]。
对一个油样进行30多组条件的试验,结果可知:当加热温度为40℃、进样量为40 pL`样品量分别为1mL,2mL和3mL时,化合物峰面积的平均相对偏差值的分布范围为0.2%~31.6%,与加热60℃和80℃时的对应值(2.9%~13.4%;1.5%~1 1.1%)相比,平均相对偏差值分布范围大,且其最大值己超过SY/T 5120-1997规定的误差范围,这可能是由于样品在40℃温度下,化合物蒸气压低,顶空进样装置中化合物的浓度过稀而使误差偏大,故样品瓶加热温度40℃不可取。
进一步选用8组不同类型化合物(直链烷烃、支链烷烃、环烷烃、芳烃),具体为2-甲基丁烷/正戊烷、3-甲基戊烷/正己烷、苯/正己烷、苯/环己烷、甲基环己烷/正庚烷、甲苯/正庚烷、二甲基环戊烷/正庚烷、三甲基己烷/正庚烷,以它们的峰面积比值来检验误差大小,结果其标准偏差都在0.03之内。这种良好的重复性说明这些比值已具备作为地化参数所需要的精度。
若采用的其他色谱条件相同,而改变样品瓶的加热温度、样品瓶中的样品量或进样量时,其结果存在一定的误差。这也提示我们,在作“油顶气”分析时,一定要注意严格控制加热温度、样品量或进样量,否则会引起较大的分析误差。
3 结果与讨论
3.1 实用性与优点
用PTV切割反吹分析法分析轻烃,能准确控制反吹时间、压力,使重组分未被汽化或汽化后尚未进入分析柱就被反吹出色谱仪,这样就减少了样品对色谱柱的污染,可延长色谱柱寿命,缩短分析周期,提高工作效率。此外,
在实验中,我们也尝试了“生物降解油”的顶空分析。因这种样品的粘度高,不能直接用注射器注入色谱仪的进样器,又因轻烃会在前处理过程中挥发掉,因此,“顶空”方法可为研究生物降解油的降解程度和生物降解对轻烃地化参数的影响提供一种新的分析手段。它具有操作流程短、省时、干扰因素少、重复性好等优点。
3.2 两种方法的对比研究
对采自“大庆”、“塔里木”、“江汉”、“吐哈”、“大港矫口“东海”6个油区的10个原油样和2-2-1项提及的9种标样分别用“反吹”、“顶空”两种方法分析,然后计算各种地化参数值及两种方法测定各种参数值的相对偏差,结果得相对偏差为±(1%~25%)。9种标样的参数值(与地化参数相同或类似的比值)
之平均值列于表2中。
-
+关注
私聊
-
yhl-87_
第105楼2009/11/03
顶空气相色谱系列讲座(100)(下)
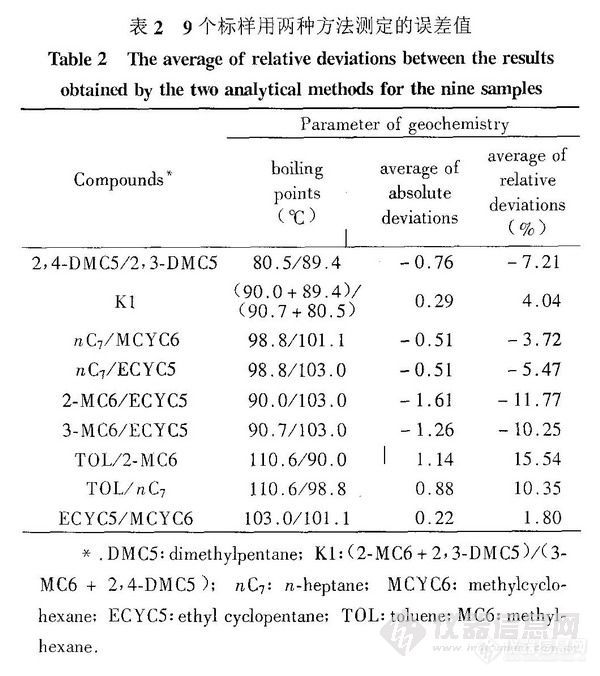
由表2可见,两种方法测定9种标样的相对偏差的平均值为-11.77%~15.54%,数值为正的是“PTV切割反吹”测定值大于“顶空”测定值,负的则反之。之所以产生偏差,显然是因“项空”分析的样品来自原油液面上部的气体,化合物在气液两相间的分配系数与化合物的极性、沸点等有关的缘故。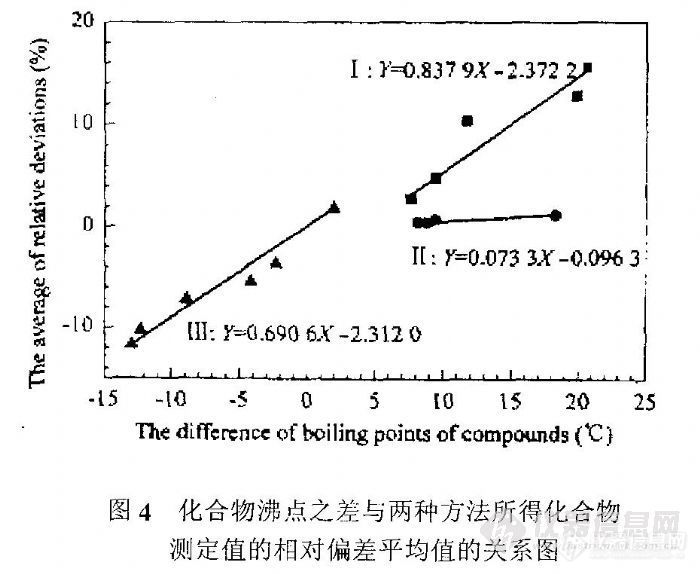
图4 化合物沸点之差与两种方法所得化合物测定值的相对偏差平均值的关系图
图4是15组标样(甲苯/正庚烷、甲苯/2-甲基己烷、甲苯/3-甲基己烷、甲苯/甲基环己烷、甲苯/乙基环戊烷、正庚烷/2,⒋-二甲基戊烷、正庚烷/2,3-二甲基戊烷、正庚烷/2-甲基己烷、正庚烷/3-甲基己烷、2-甲基己烷/乙基环戊烷、3-甲基己烷/乙基环戊烷、2,4-二甲基戊烷/2,3-二甲基戊烷、正庚烷/乙基环戊烷、正庚烷/甲基环己烷、乙基环戊烷/甲基环己烷)中每组化合物的沸点之差值与用“反吹”、“顶空”两种方法所测该组化合物的测定值(峰面积比值)的相对偏差平均值的关系图,其中的上、中、下共3条回归线分别是据第1~5组、第6~9组和第10~15组化合物所测数据得到的。
由图4可见,用“PTV切割反吹”、“项空”两种方法所得某组化合物的测定值(峰面积比值)的相对偏差平均值与该组化合物的沸点之差呈正相关,相关系数为0.89~0.99,这说明所选一组化合物的沸点之差是导致“顶空”与“反吹”测定的地化参数产生偏差的主要因素之一。此外,所选的两个化合物的类型的差别也是一个重要的影响因素,例如甲苯中芳香环会形成瞬间偶极矩,而与液相中其他化合物,尤其是极性化合物之间的分子间吸引力较大,因此,它在液相中的分配系数较非极性的链烷烃在液相中的分配系数要大,这就导致以下结果:若—组用来计算峰面积比值的两个化合物中有一个是芳烃(甲苯),则用“PTV切割反吹”和“顶空”两种方法得到的测定值的相对偏差较大,其中甲苯/2-甲基己烷的测定值的相对偏差平均值达15.54%。
4 小结
采用“反吹”、“顶空”方法分析轻烃,色谱柱寿命长,分析周期短,<C9的较轻组分的分离度能达到美国1998年ASTM D5l34-98所要求的水平,并有良好的重复性。
采用“PTV切割反吹”(或“全油”)分析方法是将原油直接注入汽化室,因此所测得的轻烃地化参数能较好的反映原油中轻烃的真实面貌;当用“项空”(或“油顶气”)的方法分析时,只将样品瓶中原油上部顶空的气体注入汽化室,因而会引起“失真”。因此,在作地化研究时,采用“PTV切割反吹”(或“全油”)色谱分析得到的轻烃有关数据的可信度高,采用“顶空”(或“油项气”)分析得到的轻烃数据要校正后才能使用。若将两类分析数据合在一起进行综合研究则更不可取。
参考文献:
[1] Mango Frank D,Org Geochem,1997,26:417-440
[2] WAl\G Pei-r°ng,ZHU Jun zhang, FANG Xiao-lin, et al Acta Petroleum Sinica, 1998, 1:24-28 王培荣,朱俊章,方孝林,等,石油学报,1998,1:24-28
[3] WU lie, LU Wan-zhen Chinese Journal of Chromatography,1984,1(I):11-17武 杰,陆婉珍,色谱,1984,1(1):11-17
[4] ZHU Jun-zhang, W A N G Pei-rong. Chinese Journal of Chromatography,1994,12(5):336~341 朱俊章,王培荣,色谱,1994,12(5):336-341
[5 ] ASTM:Designation D5l34-98
[6] SY/T5l20-1997
(全文完)
-
+关注
私聊
-
yhl-87_
第106楼2009/11/03
顶空气相色谱系列讲座(101)
固相微萃取-气相色谱法测定
食品添加剂中有害有机挥发杂质
一、前言 糖精钠、柠檬酸、苯甲酸钠是广泛使用的食品添加剂。这些食品添加剂在制造过程中可能引入微量的二氯甲烷、三氯甲烷、苯、三氯乙烯和1.4 -二恶烷等杂质。文献报道① 用顶空进样- 气相色谱法测定食品添加剂中上述5种有害有机挥发杂质,但方法所需设备昂贵,耗时较长,操作烦琐。本实验采用固相微萃取 -气相色谱联用技术(SPME-GC),测定了糖精钠、苯甲酸钠和柠檬酸中5种有害挥发杂质,并与顶空进样气相色谱法(HS-GC)进行了比较。方法操作简便、灵敏高,精密度和回收率结果能满足食品添加剂中5种有害挥发杂质定量测定要求。
本文以聚二甲基硅氧烷(PDMS)涂层的萃取纤维用来萃取糖精钠、柠檬酸、苯甲酸钠溶液中的有害有机挥发杂质:二氯甲烷、三氯甲烷、苯、三氯乙烯和1.4-二口恶烷,以HP-5毛细管柱为分离柱,火焰离子化检测器(FID)定量测定。优化了固相微萃取条件:萃取纤维、萃取方式、萃取温度、平衡时间、pH及电解质浓度等。在优化的试验条件下,进行了方法的检出限、精密度、回收率试验。5种组分的回收率在97.3%~103.9%之间。
二、 实验部分
2-1. 仪器与试剂
Agilent 6890N气相色谱仪,火焰离子化检测器(FID),Agilent 7694顶空进样器,固相微萃取装置(supelco公司),PDMS萃取纤维头100μm (supelco公司),,JENWAYP03电热磁力搅拌计,顶空瓶是20 ml。
二氯甲烷、三氯乙烯、1.4- 二恶烷(色谱纯)、三氯甲烷(色谱纯)、苯(色谱纯)、无水硫酸钠 (分析纯)。试验用水为二次蒸馏水。
2-2. 色谱条件
色谱柱:HP-5毛细管色谱柱,30 m×0.53 mm×0.25μm,进样口温度:70℃,柱温初温35℃,保持10 min,以8℃/min的升温速率升至175℃,再以35℃/min升至260℃,恒定16 min。检测器温度为260℃。
气体条件:载气为高纯氮气(N2),流速4.9ml/min;燃气为高纯氢气(H2),流速35 ml/min;助燃气为压缩空气,流速350 ml/min。
2-3. 溶液配制
① 电解质溶液的配制 称取无水硫酸钠(Na2S04)16.0 g于100 ml容量瓶中,用二次蒸馏水溶解并定容,得160g/L的Na2S04溶液。
② 标准溶液的配制 以160g/L的Na2S04溶液为溶剂,分别配制质量浓度为500 mg/L的二氯甲烷,三氯甲烷,三氯乙烯,1、4-二穗烷和苯的混合标准储备溶液,密封,于冰箱中保存,使用前用电解质溶液稀释至一定浓度。
③ 样品溶液的配制分别称取0.2 g (精确至0.0001 g)苯甲酸钠、糖精钠和柠檬酸于20 ml顶空瓶中,用10 ml电解质溶液溶解,密封保存。
2-4. 固相微萃取操作
将盛有10 ml样品溶液(或标准溶液)的顶空瓶放入40℃的恒温水浴中,恒温30 min,用SPME顶空萃取3 min,立即插入到气相色谱汽化室内进行热解吸,并进行GC分析,用外标法定量。进样顺序为标准溶液、样品溶液、样品溶液、标准溶液。样品的典型色谱图见图
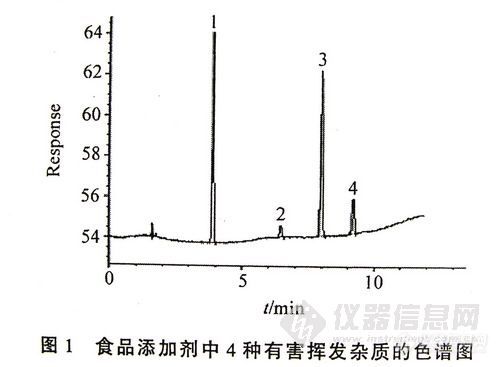
Fig.1 Chromatograms 0f the four hazardOIIs volatile impuri-
ties.m food additive samples
l一二氯甲烷;2一三氯甲烷;3一苯;4一三氯乙烯
三、结果与讨论
3-1 萃取方式的选择
固相微萃取的萃取方式主要有顶空式和浸入式两种,萃取方式的选择取决于分析物蒸汽压的大小姐],同时还要考虑待测样品基质对萃取纤维是否有损伤。对于多数的萃取涂层,含氯组分会对其造成损害,经对比试验,顶空式萃取有较好的色谱响应,且对萃取纤维的损伤较小,因此本实验选择顶空式萃取。
3-2. 萃取纤维的选择
分别用PDMS(膜厚100 tLm)和聚二甲基硅氧烷/--乙烯苯(PDMS/DVB,膜厚100肛m)两种纤维在相同的试验条件下进行顶空苹收。结果表明,PDMS纤维所得色谱响应略高且色谱峰形更为对称,因此选择PDMS萃取纤维。
3-3. 萃取条件的选择
① 萃取温度萃取温度的选择在顶空固相微萃取分析中起着重要的作用‘引。在30~80℃温度范围内,比较了温度对待测组分萃取量的影响,结果见图2。在相同的平衡时间下,40℃时萃取纤维对待测组分有较好的色谱响应,实际应用时,选择萃取温度为40℃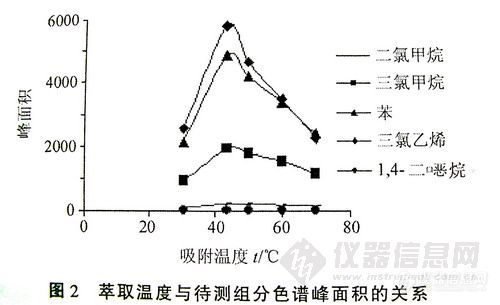
图2萃取温度与待测组分色谱峰面积的关系6492-1
Fig.2 Effect Of extraction temperature 0Ⅱthe chromato。
graphic peak areas of components
② 萃取时间 平衡时间取决于萃取纤维涂层的种类、涂层厚度、分析物的性质、搅拌速度及离子强度等¨]。实验表明随着萃取时间的增加,5种待测组分的萃取量增加很快,2 min时,萃取量即趋于饱和,此后5种组分的萃取量趋于恒定,考虑到萃取时间的选择既要保证较高的灵敏度,又要节约时间,萃取时间选择3 min。
③ 电解质的选择增大水溶液的离子强度可降低溶质的溶解度,进而增强萃取纤维对样品的萃取率。分别配制不同浓度的NaCl,Na2S04,Na3P04溶液,测定相同浓度待测组分于不同质量浓度电解质中的色谱响应,试验结果表明5种待测组分的色谱响应随着支持电解质质量浓度的增加而增大(见图3)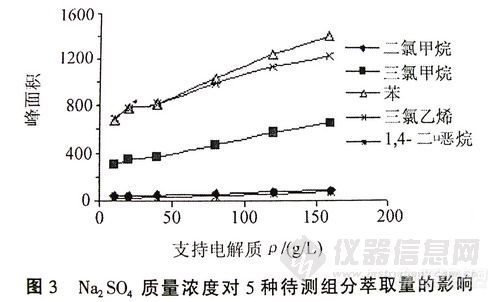
Fig.3 Effect of concentration of Na~S04 on the extraction
amount 0fthe five components
而Na2S04质量浓度的增大对五种待测组分的色谱响应的增加影响显著。但当Na2S04的量浓度过I州虬会减小扩散速度,延长达平衡的时间b。。综介考虑,本实验选取160 g/L Na2S04溶液作为离子能度调节剂。
④ 溶液pH pH对于水溶液中弱酸/弱碱化介物的电离平衡影响非常大,调整pH能增强弱酸忡,或弱碱性物质的SPME灵敏度哺’。实验结果表IlJ J:本实验中溶液pH的变化对萃取量没有显著的鼢ImpH 7时,色谱响应略高些,因此选择纯水配川IU解质溶液和样品溶液。
3-4. 方法的线性范围和检出限
在优化萃取条件下进行模拟样品中二氯甲烷、三氯甲烷、三氯乙烯、1、4--"砥烷和苯5种有害挥发杂质的测定。5种待测组分的线性回归方程、线
性相关系数、线性范围和检出限见表1。
3-5. 方法的回收率和精密度
取苯甲酸钠,糖精钠和柠檬酸样品溶液,按线性范围分别添加不同质量浓度的标准溶液,重复测定16次,回收率和精密度实验进行结果见表2。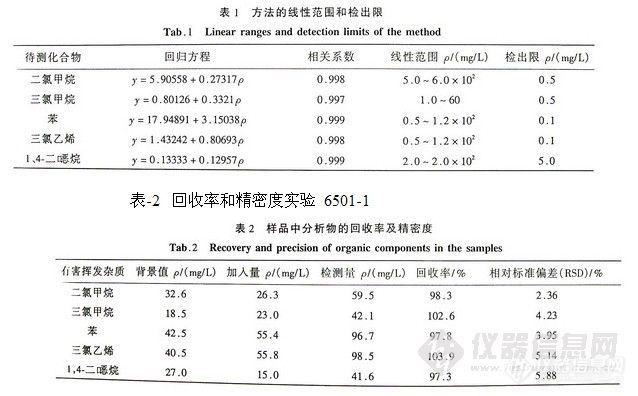
3-6. 与顶空气相色谱法的比较
分别配制了系列浓度的二氯甲烷、三氯甲烷、苯、三氯乙烯和1.4-二恶烷的标准溶液,分别对5种待测组分进行顶空气相色谱(HS-GC)实验和SPME-GC测定,以比较两种样品富集和前处理方式的优劣。结果表明与顶空法比较,固相微萃取样品富集和前处理技术对挥发性较强的二氯甲烷、三氯甲烷和苯具有较高的色谱响应,而对三氯乙烯和1.4-二恶烷两种实验方法色谱响应相差无几。与顶空气相色谱法相比固相微萃取具有简便、灵敏、回收率高等优点。
参考文献
[1] U S Standard. Number:USP24,Oganie volatile impurities (467),2000
[2] 肖珂,王勇,路鑫等.色谱,2003,21(1):76
[3] 杨 敏,仇文丽,曾昭睿待.分析试验室,2002,27(21):2002
[4] 赵汝松,柳仁民,崔庆新.分析化学,2002,30 (6):722
[5] Buchlolz K D,Pawlixzynm J.Anal Chem,1994,66:160
[6] 路鑫,赵欣捷,叶芬等.色谱,1999,17 (2):131
-
+关注
私聊
-
yhl-87_
第107楼2009/11/03
顶空气相色谱系列讲座(102)
药用丁基橡胶塞中易挥发性成分的顶空分析
摘要 目的:分析医药中使用的丁基橡胶塞的挥发性成分。
方法:采用气相色谱-质谱法(GC-MS)和NIST谱库分离鉴定。
结果:药用丁基胶塞中主要检出21种挥发性成分,其中硅烷化试剂、抗氧剂、饱和烷烃类化合物在药用丁基胶塞的挥发性成分中所占的比例相对较高。与普通丁基胶塞、氯化丁基胶塞、溴化丁基胶塞和镀膜丁基胶塞所含挥发性成分存在一定的差异。
结论:药用丁基胶塞中挥发性成分的分析,为进一步进行药用丁基胶塞与药物之间的相容性考察,奠定了基础。
本文是中国药品生物制品检定所的赵霞、胡昌勤和金少鸿的课题的部分内容,这也是顶空气相色谱分析方法在各种有机材料方面的应用,现全文介绍如下:
前言
药品是一种特殊的商品,其药效与质量直接关系到人身健康和安全,药品的包装材料与结构形式,尤其是直接接触药品的包装材料,对保证药品稳定性起决定性作用。不适宜的包装材料可引起活性药物成分在包装材料上的吸附,或者吸附包装材料中的成分,甚至发生化学反应,导致药品失效,有时还会产生严重的毒副作用。因此,药包材的选择是否合适,是药品质量评价的一项重要指标[1]。
丁基胶塞是药品包装中瓶装密封材料的重要组成部分,具有吸湿率低,化学性好,气密性好,无生理毒副作用等显著特点,特别适于用作药品密封,为药物质量提供了更为可靠的保证。但胶塞的制造还必须加人多种化学助剂,且需要经过硫化过程,因而不可避免的会在胶塞本体内残存一些未被化学键合的反应剩余物,不参与反应的物质、热分解产物和硫化生成物。在胶塞的贮存过程中,尤其是在高温灭菌(一般灭菌条件为121℃灭菌30 min)时,这些物质不可避免的会迁移到胶塞表面,而影响或者污染药物。从市场反馈的信息来看,某些品种的抗生素注射用粉针制剂在贮存过程中,存在溶液澄清度超标的现象,如注射用头孢曲松钠、头孢唑林钠、头孢噻肟钠等,均怀疑可能与所用胶塞质量有关。为此,胶塞制造企业为隔离瓶塞与药品的相互接触,已陆续研制和开发出镀膜胶塞和涂膜胶塞,即在胶塞表面或与药物接触面采用不同的工艺涂覆一层四氟乙烯、聚乙烯或聚丙烯等材料膜。由于这些材料优越的化学惰性,使得胶塞与药品的相互反应几乎降低到了最低限度。截止到目前,这是解决瓶塞与药品相容性问题最有效的方法。但由于该类胶塞的制造成本较高,售价要比常规胶塞高出很多倍,因而应用范围还相当有限。因此如何将迁移物降到最低的程度,从而研制出更高档次的丁基胶塞,是当前国内外不断探索和研究的新课题。
作为药物的包装材料,胶塞中所含的易挥发性成分迁移到药物中,或者被药物吸附的可能性比较大,但由于受到分析仪器的限制,对胶塞中挥发性成分研究的国内外文献并不多见。近年来,随着顶空气相色谱、毛细管气相色谱以及气相-质谱联用技术的发展,对胶塞成分的分析成为可能。Fa Zhang[2]等人利用GC-MS技术和有机合成技术,对胶塞的乙腈提取液中的4种成分进行了结构确证;N.Delaunay-Bertoncini[3]等人将胶塞剪碎,利用顶空和索氏提取对胶塞中的成分进行了分析。但他们均是将胶塞剪碎,破坏了胶塞在使用过程中的形态,不能真实的模拟丁基胶塞作为包装材料,在与药物接触过程中,胶塞中的成分,特别是挥发性成分的迁移过程。在本文中,将完整的丁基胶塞作为研究的对象,利用顶空气相色谱和气相-质谱联用技术,对药用丁基胶塞(包括普通丁基胶塞、氯化丁基胶塞、镀膜丁基胶塞和溴化丁基胶塞)中的易挥发性成分加以分离,并利用NIST谱库,对丁基胶塞中的多种挥发性化合物进行了分离鉴定,为进一步研究丁基胶塞与药物的相容性奠定了基础,国内尚未见报道。
实验
1 仪器与试药
Thermo Finnigan Trace GC/Polaris Q气-质联用仪;Mettler电子天平;丁基胶塞1(批号0305027):由重庆三海兰陵公司提供;丁基胶塞2(批号0312054-硅烷化等级I级):由郑州市嵩山企业集团翱翔医药包装有限公司提供;氯化丁基胶塞1(批号0310052057):由山东药玻橡塑包装材料分公司提供;氯化丁基胶塞2(批号0309081-09):由江阴兰陵瓶塞公司提供;镀膜丁基胶塞:由山东瑞阳制药有限公司提供。溴化丁基胶塞:由国外某胶塞厂提供。
2 方法与结果
2.1 GC-MS的色谱条件
顶空条件:121℃顶空30 min;色谱柱:DB -1MS毛细管气相色谱柱,0.25 mm×30 m;载气:高纯氦气,流速1,0 mL/min;柱温:50℃,保持5 min,以5℃/min速度升温到200℃,保持6min。进样体积:1 mL.
2.2 GC-MS的质谱条件
电子轰击离子源(El),轰击电压70 ev,离子源的温度为200℃,接口温度200℃,质量分辨率1.0 amu;全扫描方式,扫描的质量数范围50~650,工作站是Xcalibur。
2.3 顶空样品的制备
将一整胶塞放置于20mL的顶空瓶中,加相同胶塞密封,并加铝盖封严。
2.4 实验结果
按上述实验条件对药用丁基胶塞的挥发性成分进行GC-MS分析,总离子流图见图1。用气相法分离并经质谱扫描后主要检出21个色谱峰及对应的质谱,经Xcalibur工作站中的NIST质谱库检索,并结合有关文献人工图谱解析鉴定,共鉴定出11个成分,结果见表1。各化合物的峰面积相对含量分析是通过Xcalibur 工作站的数据处理系统,按峰面积归一化法进行,结果见表2。

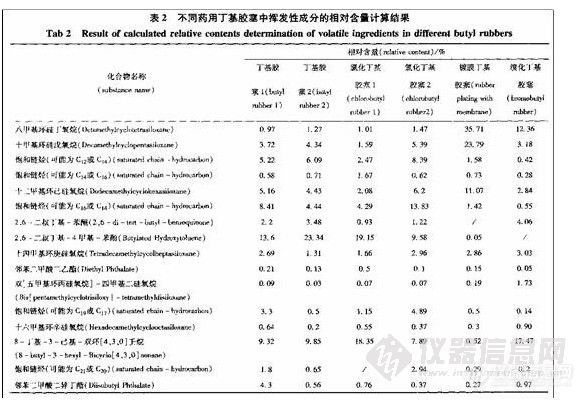
3 讨论
3.1 分析结果表明,丁基胶塞中的挥发性成分主要可以分为三类,第一类主要是环硅氧烷类化合物{[Si0(CH3)2]n},如保留时间为~12.35 min;~17.54min;~22.50 min等的谱峰,n的取值主要从4~8。各硅氧烷化合物的归一化含量的总和,丁基胶塞1为13.18%;丁基胶塞2为1 1.55%;氯化丁基胶塞1为6.89%;氯化丁基胶塞2为16.39%;镀膜丁基胶塞为73.73%;溴化丁基胶塞为22.31%。第二类主要是在胶塞合成过程中添加的抗氧剂,如保留时间~26.10rnin的谱峰为2,6-叔丁基-4 -甲基-苯酚(BHT)丁基胶塞1、丁基胶 9.58%,镀膜丁基胶塞中的BHT含量非常微量,溴塞2、氯化丁基胶塞1和氯化丁基胶塞2中BHT的归一化含量分别为13.60%;23.34%;19.15%;, 化丁基胶塞中没有发现BHT。第三类就是饱和烷烃类,如保留时间~18.36 min;~23.87 min;~28.70 min;~31.08 min的谱峰,但是从NIST谱库中不能确定饱和烷烃的碳原子数目。
-
+关注
私聊
-
yhl-87_
第108楼2009/11/03
顶空气相色谱系列讲座(102) (下)
3.2 环硅氧烷类化合物是胶塞进行硅烷化处理时,需要加人的硅油成分,也是胶塞总离子流(TIC)图中所占比例最大的一类化合物。胶塞硅化的目的是在胶塞表面涂上一层硅膜,以便胶塞的分装和压塞。常规瓶塞后处理均需要进行硅化,防止在贮存过程中发粘,使用过程中能很好的上机压塞,并且还可以防止在运输过程中减少表面的摩擦,避免摩擦造成胶屑微粒,所以硅化是目前胶塞生产厂家的后处理工艺[4]。目前,胶塞生产企业按每平方厘米比表面积甲基硅油含量的毫克数将硅化分为A,B,C三个等级,其硅化丁基指标分别为:A级0.01~0.03 mg/cm 2,B级0.03~0.06 mg/cm2,C级0.07~0.12 mg/cm2[5]。硅油虽然是一种惰性材料,但对其药液浊度影响很大,因为胶塞在与药粉接触后很容易被硅油所吸附,形成胶体或者药粉团,胶塞接触面积越大,所形成的胶体或者药粉团也就越多。药品稀释后因硅油本身就不溶于水,所以容易出现遇水产生浑浊和药粉难溶的现象。另外,输液产品在经过120℃(1~3 h)的高温灭菌,冷却到常温后,会发现瓶子上有“挂珠”现象,因为胶塞表面的硅油在高温时分子热运动加剧,胶塞表面的硅油脱落后附于瓶壁而造成。为避免影响到药物的质量,胶塞表面的硅化程度应该因药而异[4]。在所考察的胶塞中,环硅氧烷类化合物在挥发性成分中的归一化含量均是比较高的。
3.3 在普通丁基胶塞和氯化丁基胶塞的挥发性成分中归一化含量相对较高的2,6-叔丁基-4 -甲基-苯酚(BHT),是胶塞工业中比较常用的一种抗氧剂。但在溴化丁基胶塞中没有发现该抗氧剂,这主要是因为酚类抗氧剂对溴都比较敏感,一般用其它抗氧剂所代替,如环氧化豆油。
BHT是一种酚类抗氧剂,主要用于橡胶、塑料等加工工业,也作为油脂抗氧剂使用。今年来,国外不少学者研究发现,BHT对肺、肝等组织有直接损害作用,并有促进肿瘤发生的可能。国内尚未见有报道[7 8]。目前在日本、欧洲、美国已禁止BHT在婴儿和儿童食品中使用。因为在有关国际会议上曾对BHT的评价认为:虽没有发现基因毒性和致癌性,但它是潜在肿瘤促进剂,对它的长期影响还需进一步研究。
从各胶塞的GC-MS总离子流图中可以看出,国内生产的普通丁基胶塞与氯化丁基胶塞所含有的挥发性成分基本一致,但化合物的种类相对于溴化丁基胶塞和镀膜丁基胶塞来说,明显增多。
所考察的镀膜丁基胶塞的总离子流图谱中除了存在明显的环硅氧烷化合物外,其它挥发性成分相对较少,这显示了胶塞表面镀惰性膜的作用。
4 结论
本文利用GC-MS对丁基胶塞的挥发性成分进行了分析,共分离出21种化合物,并对其中11种化合物进行了结构确证,为进一步研究胶塞与药物的相容性奠定了一定的基础。
某些抗生素品种,如注射用头孢曲松钠,在其溶液澄清度不合格的样品中也发现存在一种或几种来自药用丁基胶塞挥发性成分的微量杂质。从药用丁基胶塞释放到药物中的这些药用丁基胶塞的挥发性成分是否会影响到药物溶液的澄清度、疗效,甚至引起药物的毒性,以及是否将药物中所含源自药用丁基胶塞的挥发性成分作为评价药物质量的一个指标,有待进一步的试验证明和科学探讨。
参考文献
1 LI Yong-an(李永安) Applied manual of pharmaceutical packaging(药品包装实用手册)Beijing(北京):Publishing House of Chemical Industry(化学工业出版社),2003:27
2 Zhang F,Chang A,Kamisz K,et al.Structural identification of extractables from rubber closures used for pre-filledsemisolid drug applicatior by chromatography,mass sepectrometry and organic synthesis J Pham Bomed Anal. 2004,34(5):841
3 Delaunay-Bertoncini N,van der Wielen FW,de Voogt P,et al Analysls of low – molar- mass materials in commercial rubber samples by Soxhlet and headspace extractions followed by GC-MS analysis J Pharm Biomed Anal 2004,35(5):1059
4 WANG Fei(王飞),SI Jun -Jie(司俊杰),Butyl rubber free of silylation and particulate for medical use(免硅化无微粒的医用丁基橡胶瓶塞).Auto- electricity Inform(机电信息)2004,21:35
5 HU Xu - mao(胡绪茂),HUA Guo-ping(华国平),Factors influencing machining probability of butyl rubber(影响丁基胶塞上机率的各种因素)Chin Pharmacy(中国药业)2003,12(3):38
6 ZHOU Shu Dan(周树南) Fatalness evaluation and development of food additives(食品添加剂的危险性评估与发展)Jiangsu Health Care(江苏卫生保健)2001,3(3):6
7 Powell C J,Connolly JC,Jones S M,et al Hepatic responses to the administration of high doses of BHT to the rat:their relevance to hepatocarcinogenicity.Food Chem Toxicol. 1986,24(10 - 11):1131
8 Kehrer J P. Blenmycin and cyclophosphamide toxicity In mice with damaged lung tissue.Toxicology.1989,57(1):69
(全文完)
-
+关注
私聊
-
yhl-87_
第109楼2009/11/03
顶空气相色谱系列讲座(103)-土壤中挥发有机物
1 前言
挥发性有机物对土壤的污染,在发展中国家这种现象越来越突出。有机溶剂的泄露、处理不当等均可导致对土壤和水的污染。而VOCs(挥发性有机物)具有迁移性、持久性和毒性,是一类重要的环境污染物.对人体具有致畸、致突变和致癌等作用。环境样品中残留的各种挥发性有机物的含量都很低,要高效准确地分析土壤中挥发性有机物一直为国内外所关注。.上世纪70年代美国环保局将这些挥发物VOCs的分析技术应用于土壤基质。但大量研究报道表明,对土壤采用洗提和提取手段,实际的回收率并不高,而且手工操作一方面易造成有机溶剂的消耗.另一方面也易导致待测土壤中VOCs的损失。针对这个问题,现在美国对于环境基质中VOCs的分析多采用液上顶空 (HS)、吹扫、捕集、洗提等分析方法,自动样品处理的顶空技术被应用到VOCs的分析中,可避免样品转移过程中的损失。Eitzer ⑴、Tolval1en 等⑵ 和Wilkins⑶用顶空技术测定了固体废物中的烷基苯(甲苯、乙苯、萘、异丙基苯、丁基苯)。对固体样品VOCs的分析有多次顶空提取⑷⑸ 和顶空-固相微萃取(HS-SPME)⑹。美国EPA对土壤中VOCs前处理方法存在于EPA 5000系列中,
VOCs前处理方法主要有:溶剂萃取并直接进样(高浓度样品)、顶空分析(EPA5021)、吹扫捕集(EPA5030B).密闭系统吹扫捕集(EPA5035)。多年来,国内对大气和水体中的VOCs报道较多,在国家环保总局编制的监测方法中就提供了相应的分析方法⑺⑻。但国内对于土壤、沉积物中VOCs测定的前处理技术标准方法尚未建立,所以研究通过顶空前处理技术测出土壤、沉积物受VOCs的污染状况,具有实际意义。
本文由鞍山市环境监测中心站的孙华和田靖两人重点参考美国的EPA方法,寻找选择适合于土壤中挥发性有机物提取的顶空条件,用HS-GC/FID来对土壤中VOCs进行分析,并做了准确度和精密度的评价。
2 材料和方法
将样品置于顶空瓶内,样品中的挥发性组分就会向容器的液上空间挥发.产生蒸气压,在一定条件下,当气液两相间达到热力学动态平衡时,取气相样品进行色谱分析。用液上空间法制备样品.实质上是把存在于水相中的目标化合物转移至气相.通过对液上空间气体的分析.从而完成对样品的测定。该法具有操作简便、分析快捷、准确灵敏、干扰少等特点。本方法适用于在顶空平衡程序中有较好挥发性的有机物,因此适用范围较广。用适合EPA方法的含54种VOCs标样配制一标准溶液,用此标液做出一工作曲线。标准样用甲醇制得,工作曲线样品则是将标准样加入到含10mL基质修正液的顶空瓶中。基质修正液是用磷酸调成pH为2的NaCL饱合溶液。取加标土壤样品2g于22rnL顶空瓶中,加入10mL基质修正液,再加入5uL标准VOCs,使土壤VOCs含量为50μg/kg,然后用PTFE/丁基的铝盖将顶空瓶迅速盖好。顶空瓶在振荡器中振荡10min.然后放入顶空装置中。完成上述程序后.按照表1所列条件进行顶空处理和样品的气相分析。
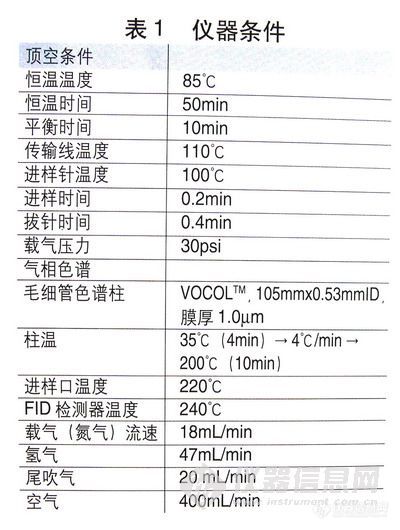
3 结果和讨论
应用顶空前处理技术,选择合适的气相条件,完全可以满足对土壤、沉积物中挥发性有机物的提取和分析的要求。在顶空前处理下的VOCs的色谱图见图1。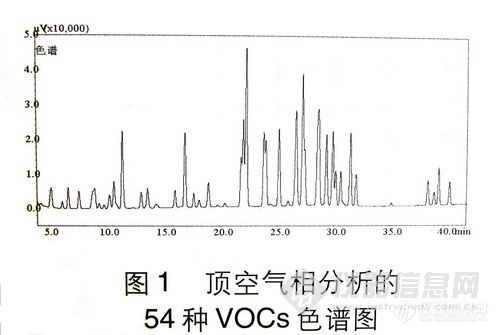
3.1 顶空密封垫的选择
用同一种浓度的54种VOCs标准溶液对四种不同材料的顶空瓶密封垫进行选择.分别是丁基、铝/硅氧烷、PTFE/硅氧烷和PTFE/丁基,从VOCs的GC响应结果表明,后三种密封垫明显好于丁基材料,而PTFE/丁基的密封垫被证明是最佳材质的密封垫。
3.2 基质修正液及加入量的选择
基质修正液的加入可以消除或减少基质效应的影响。
3.2.1 利用盐析作用.即在样品中加入无机盐(氯化钠)和水的方法,可以改变挥发性有机物的分配系数,通过进一步地调节pH,使待测物的挥发性更大。因此在对土壤样品进行HS-GC分析,需要在土壤样品中加入一定量的基质修正液,从而有利于有机物从土壤中脱附出来。参考EPA5021,用H3PO4调500mL无有机物的水至pH等于2,再加180g NaCI,配成为显酸性的氯化钠饱和溶液。
3.2.2 取2g土壤样品于顶空瓶中.并向其加入一定量标准液.再向此顶空瓶中分别加入不同量的基质修正液:0.6mL、2mL、5mL、8mL、9mL、10mL,结果表明.加入0.6mL基质修正液(即水/土比30%左右)时效果最好,而其它加入量基本相差不大,其结果见图2。这一点与肖锐敏等《项空气相色谱法测定土壤中BTEX》一文中所作的分析一致。但考虑EPA5021中10mL基质修正液的加入量,及对土壤样品在顶空瓶中的覆盖程度,忽略基质的影响而最终简化成气液两相的关系,本实验最终采用基质修正液的加入量为10mL。
3.3 顶空平衡温度的选择
样品的平衡温度与蒸汽压直接相关,它影响分配系数。一般情况下温度高则蒸气压高,顶空气体的浓度就高,分析的灵敏度相应也提高,因此平衡温度较高,对低沸点的挥发性有机物在顶空气相中的分析具有一定好处。取2g加有标准物的背景土于顶空瓶中,分别在20℃、30℃、40℃、50℃、60℃、70℃、80℃、85℃、90℃,根据结果及考虑挥发性有机物在较高温度下对固-液一气三相的平衡关系的改变.并结合国内外的文献资料,平衡温度选定为85℃。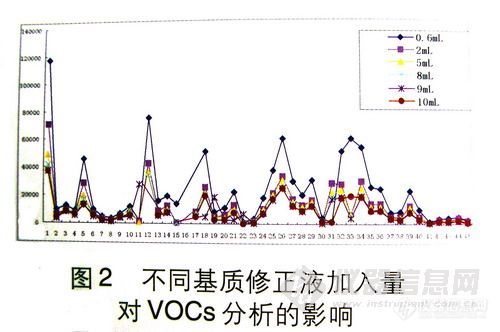
3.4 顶空平衡时间的选择
平衡时间主要由被测组分分子从样品基质到气相的扩散速率所决定。在平衡温度85℃的条件下.平衡时间分别设定为10min、20min、30min、40min、50min、60min,结果选平衡时间为50min。
3.5压力化时间的选择
在上述选定的条件下.压力化时间分别设为2min、5min、10min、20min、30min,取液上气体进行测定.选10min为压力化时间。
3.6 进样时间的选择
进样时间决定进入GC中的进样量.由于采用的是自动顶空进样系统,所以进样量的重现性可以保证。在上述顶空条件下.分别设定进样时间为0.02min、0.05min、0.1min、0.2min、0.3min、0.4min,GC的分析结果证明0.2min是最佳的进样时间。
3.7 加标率测定
用顶空处理技术,根据多次实验回收率的分析最好采用两种基质:沙子和花园表层土,这一结论与EPA8260中的结论相同。花园土挥发性有机物的加标量为50μg/Kg,平均回收率为84.1%。
4 结论
应用顶空技术对土壤、沉积物样品进行前处理,能够最大程度地避免VOCs的挥发损失。选择本方法的顶空条件,可以保证液上空间有最佳的待测组分,从而满足GC对VOCs分析灵敏度的要求,此方法操作简便、具有可行性。
摘自perkinElmer公司的《China Link》2006年第二期
-
+关注
私聊
-
yhl-87_
第110楼2009/11/03
顶空气相色谱系列讲座(104)-大孔吸附树脂在前处理中带入的残留溶剂
顶空气相色谱法测定三七三醇皂苷中
因大孔吸附树脂带入的有机残留物
摘要 目的:采用顶空气相色谱法测定三七三醇皂苷中因大孔吸附树脂带人的正己烷、苯、甲苯、二甲苯、苯乙烯和二乙基苯类残留量。
方法:采用Zebron ZB-Wax弹性毛细管(30 m×0.53 mm×1.0μm)色谱柱,氢火焰离子化检测器(FID),程序升温,进行顶空气相色谱测定。
结果:6种有机溶剂完全分离,在所考察的浓度范围内相关系数均大于0.99以上;回收率为85.7%~101.3%;检出限为0.1~1.1μg·mL-1;精密度为RSD为1.38~5.13%。
结论:该方法简便易行,测定结果准确可靠,可用于三七二醇皂苷中大孔吸附树脂残留物的测定。
本文由四川省食品药品检验所的王野、杨蕾、袁军、刘仲义研究人员共同完成,现全文介绍如下
前言
三七三醇皂苷生产过程中虽未使用一、二类有机溶剂,但富集纯化过程中曾使用大孔吸附树脂。按照国家药品监督管理局关于大孔吸附树脂有机残留物的规定[2],需建立药品中大孔吸附树脂残留物“正己烷、苯、甲苯、二甲苯、苯乙烯和二乙基苯类”残留量的测定项目和方法。本文首次采用顶空气相色谱法同时测定6类大孔吸附树脂有机残留物,获得了重现性好、灵敏度高的分析结果。
实验
1 仪器与试药
HP 4890型气相色谱仪及工作站,HP 7694E顶空进样器。三七三醇皂苷由成都希臣药业有限责任公司提供;正己烷为色谱纯;苯、甲苯、二甲苯、N,N一二甲基甲酰胺(DMF)为分析纯;苯乙烯和二乙基苯类在国内外均无试剂和标准品,我们选用的是符合国家标准[7]的工业用原料。
2 实验方法
2.1 色谱条件
色谱柱:Zebron ZB -Wax毛细管色谱柱(30 m×0.53 mm×1.0μm),柱温采用程序升温:柱温60℃,保持5 min,再以8℃·min-1升温至150℃并保持5 min;进样口温度:200℃;FID检测器温度220℃;氮气为载气,流速l0 mL·min-1。顶空条件:顶空平衡温度60℃,平衡时间50 min,进样环温度120℃,进样体积1 mL。
2.2 溶液的制备与测定结果
标准储备溶液:分别精密称取正己烷、苯、甲苯、二甲苯、苯乙烯、二乙基苯类适量,置50 mL量瓶中,用DMF溶解并稀释制成0.2 mg·mL-1(正己烷2.9 mg·mL-1、甲苯8.9 mg·mL-1、二甲苯21.7 mg·mL-1、苯0.02 mg·mL-1)的溶液。精密量取该溶液2.5 mL,置50 mL量瓶中,用水稀释制成标准储备溶液。
标准溶液:精密称取样品1 g,置顶空取样瓶中,加人2 mL标准储备溶液,密封,超声混匀,作为标准溶液。
供试品溶液:精密称取样品1 g,置顶空取样瓶中,加入2 mL 5%DMF溶液,密封,超声混匀,作为供试品溶液。
大孔吸附树脂溶液:分别称取未经处理和已处理的大孔吸附树脂2g ,置顶空取样瓶中,加水2 mL,密封,超声混匀。
空白溶液:2 rnL 5%的DMF水溶液,置顶空取样瓶中,作为空白溶液。该色谱条件能将各个残留溶剂分离,5%DMF水溶液对以上各种有机溶剂的检查无影响(见图1)。未经处理和已处理的大孔吸附树脂溶液测定图谱见图2。

3 方法与结果
3.1 检测限的测定
精密吸取“2.2”项下标准储备溶液适量,用水准确逐级稀释,直至其峰高约为噪音的3倍,结果正己烷、苯、甲苯、二甲苯、苯乙烯和二乙基苯类六种最低检出浓度分别为1.1,0.1,0.2,1.1,0.8,0.9μg·mL-1。
3.2 进样精密度试验
精密量取同一标准溶液(正己烷、二甲苯、苯乙烯、二乙基苯类、苯和甲苯浓浓度分别为13.20, l 1.80,l 1.20,12.10,0.95,10.35μg·mL-1)的顶空气体各1 mL注人气相色谱仪,连续进样5次,记录色谱图,量取峰面积,结果RSD分别为5.13%,4.65%,3.31%,2.64%,1.38%,1.39%(n=5)。
3.3 线性关系试验
分别精密量取系列浓度的正己烷、苯、甲苯、二甲苯、苯乙烯、二乙基苯类的顶空气体各1 mL注人气相色谱仪测定,以浓度为横坐标,峰面积为纵坐标,绘制标准曲线,结果见表1。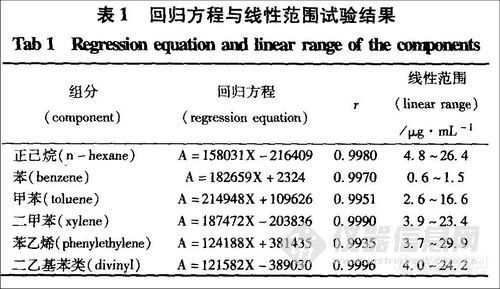
3.4 重复性试验
按样晶测定方法,取三七三醇皂苷样品(批号031201)5份进行测定。试验结果除苯的百分含量的平均值为0.00013%;RSD为2.4%,其余溶剂未检出。
3.5 回收率试验
精密称取三七三醇皂苷样品(批号031201)1 g,加入标准储备液2 mL溶解,气体1 ml,自动进样。另取一份样品、标准储备液按2.2方法制备后测定。根据峰面积比计算回收率正己烷、苯、甲苯、二甲苯、苯乙烯、二乙基苯类的平均回收率(n=5)分别为99.2%,85.7%,92.4%,95.2%,98.1%,101.3%。
3.6 样品的测定
取样品3批,照“2.2”项制备标准溶液和供试品溶液,采用标准加人法计算各样品中大孔吸附树脂的残留物量。3批样品(031201、031202、031203)除苯的残留量分别为0.00014%,0.00013%,0.00012%外,其余均未检出。
4 讨论
4.1 色谱条件的选择
大孔吸附树脂目前国内外尚无残留物的规定标准,上述残留溶剂中正已烷、二甲苯、苯和甲苯的检验方法及限度在2005年版中国药典二部附录Ⅷ P [1] 己有收载,而二乙基苯类和苯乙烯的限量及检验方法尚无法定标准收载,但其分析方法在国家标准GB/T 12688.1 - 1998[6] 及SH/T1485.2 - 95[ 7] 中有收载。实验中曾用药典方法的色谱条件试验,结果苯、甲苯的分离均符合要求,但其余有机残留溶剂的分离达不到要求。后选用极性毛细管色谱柱,采用程序升温顶空气相色谱法,结果6种有机溶剂均达到了完全分离。其中二甲苯是三个相互未分开的混合物,按邻、间、对二甲苯二个异构体的面积和定量;二乙基苯类包括邻、间、对二乙基苯和萘[3][7],同样按4个峰面积的和定量。
4.2 供试品溶剂的选择
实验中先选用水作溶剂,结果苯乙烯在水中难溶,经试验,采用对多种化合物有溶解能力的二甲亚砜和二甲基甲酰胺作溶剂,试验结果表明:二甲基甲酰胺较理想。由于测定对象均为非极性物质,样品本身易溶于水,故选择DMF:水的比例为(5:95),既能保证样品的溶解,又不至DMF蒸汽压过高,影响检测灵敏度。
4.3 定量方法的选择
微量物质的分析常见定量方法有外标法、内标法和标准加人法[5]。根据顶空进样的定量原理,一般用内标法,但要寻找出一个与6种残留溶剂结构均相似的内标物质显然较困难。因此,我们选用了标准加入法,本次实验中在相同重量的样品中,一份加溶剂溶解样品作为供试品溶液,另一份加人相同体积的欲测组分的标准储备溶液作为标准溶液,在相同色谱条件下,测定加人标准储备溶液前后欲测组分的峰面积,可以计算出原样品中欲测组分的含量。
-
+关注
私聊
-
yhl-87_
第111楼2009/11/03
顶空气相色谱系列讲座(104)(下)-大孔吸附树脂在前处理中带入的残留溶剂
4.4 残留溶剂的归属
在未处理的大孔吸附树脂中检出了10种主要有机溶剂,其中8、9号峰为二乙基苯类,其余峰未定位,但经过处理后的大孔吸附树脂仅检出微量的3种有机溶剂,其中3号峰为苯,未检出其他5类已定位的残留有机物,这与三七三醇皂苷样品测定的结果一致(见图-2)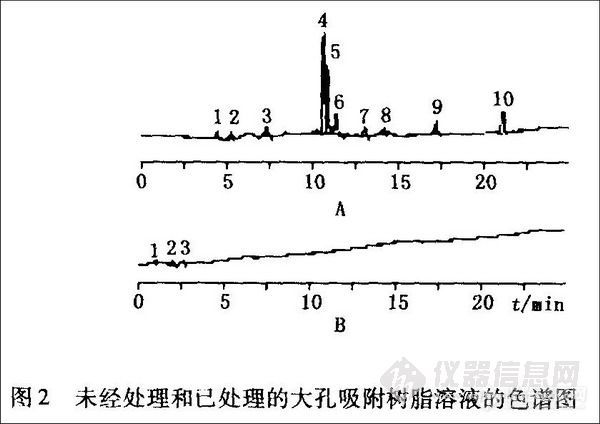
A 未处理过的树脂 B 已处理过的树脂
说明大孔吸附树脂经过严格的前处理确实可以除去挥发性残留物[4],而且三七三醇皂苷中残留的苯可能由已处理的大孔吸附树脂引人。
4.5 限度的规定
在药审中心关于“大孔吸附树脂分离纯化中药提取液的技术要求”的补充说明中,暂定为苯< 2 ppm、甲苯< 890 ppm、二甲苯< 2170 ppm、正己烷、苯乙烯和二乙基苯类其限量不能高于国际标准或国际通用标准。结合实际生产中大孔树脂用于该药纯化工艺时,经过了严格的前处理,所以三批样品的残留物除苯很低外,其余均未检出,故参照2005年版中国药典二部附录Ⅷ P [1] 将苯、甲苯、二甲苯、正己烷、苯乙烯和二乙基苯类的残留物限度定为正己烷小于290 ppm,苯小于2 ppm,甲苯小于890 ppm,二甲苯2170 ppm,苯乙烯和二乙基苯类参照国家标准[5] [7] 及实验测定结果定为小于20 ppm。
参考文献
1 Chp(中国药典),2005 Vol Ⅱ(二部〉:Appendix(附录)Ⅷ P
2 National center for Drug Evaluation(国家药品审评中心)Guidance on Macroporous Crosslinked Polystyrenes Used to Purify Extracts of Chinese Traditional Medicinal Products(Interim)[大孔吸附树脂分离纯化中药提取液的技术要求(暂行)]
3 JIN Zhang-zhao(金樟照),WU Wen-Jun(吴文军),ZHU Ming(祝明)Identification of Organic Residues from Four Types of Macroporous Crosslinked Polystyrenes by GC/MS(4种聚苯乙烯型大孔吸附树脂有机残留物GC/MS分析).Chin J Pharm Anal(药物分析杂志),2004,24(6):647
4 LING Ning-sheng(凌宁生),LIU Zhi - qing(刘志清),LI Lin(李林),et al Analysis of benzene - series residues In d - 101 macroreticular resin used for TCM(中药用D-101型大孔吸附树脂苯系列残留物分析研究)Chin Tradit Herb Drugs(中草药),2002,33
(2);122
5 LIANC Han - chang(梁汉昌).Trace Substantial Analysis by Gas Chromatography(痕量物质分析气相色谱法)Beijing(北京):China Petrochemical Press(中国石化出版社),2000. 5
6 China Standard(中华人民共和国国家标准)CB 3915 - 1998,CB/T 12688 I - 1998
7 China Petroleum and Chemical Industry Standard(中华人民共和国石油化工行业标准).SH/T 14851 -95,SH/T l4852 - 95
-
+关注
私聊
-
yhl-87_
第112楼2009/11/03
顶空气相色谱系列讲座(105)(上)
自制SPME顶空装置和复合固相微萃取涂层的研究与应用
摘要: 以苯乙烯-丙烯酸乙酯共聚物与色谱固定液SE-30结合作为固相微萃取头涂层。考察了复合涂层的热稳定性及其与纤维结合能力,自制了SPME装置。采用HS-SPME-GC技术萃取分析了水中一氯苯、二氯苯、三氯苯。以色谱峰高对浓度作外标曲线,在10-l00mg/L范围内,一氯苯、二氯苯、三氯苯的线性相关系数分别为0.9968,0.9976, 0.9904,最低检测限分别为0.32 mg/L、0.27 mg/L, 0.50 mg/L。 样品测试中,相对标准偏差(n=5 )分别为5.37%, 4.78%, 6.63% ,加标平均回收率分别为94.4%, 96.7%, 94.2%。 将自制涂层与商品涂层(PDMS, PA)对水中氯代芳烃化合物萃取量进行了比较,结果表明苯乙烯丙烯酸乙酯聚合物(SE-30复合涂层)对芳烃化合物具有优良的吸附特性。
1 前言
固相微萃取(SPME)是一种新型的样品前处理技术⑴ ⑵,是通过吸附、溶解等作用将被萃取物浓缩于涂层中,然后通过高温或溶剂洗脱方法将被分析物解析下来,分析目标物。适用于SPME涂层的色谱固定液数量有限。目前,商品化的SPME装置只有美国Supelco公司生产的涂有聚二甲基硅氧烷(PDMS),聚丙烯酸酯(PA)和聚乙二醇2万(PEG20M)3种单一吸附质及4种部分交联的复合固相涂层材料。Supelco公司生产的SPME装置价格贵,石英纤维易碎,一旦纤维破碎或折断,整个萃取头便作废了。在使用中,弹簧内萃取头后部的金属丝在推拉时易弯曲折断,涂层也经常出现脱落现象。SPME涂层极性不同其应用范围不同。因此,开发新型涂层、改进SPME装置、扩大加强联用性是SPME技术当前的主要研究内容。近几年,非商品化的涂层已经发展到了10种左右⑶。商品化的SPME涂层的应用研究已有较多报道⑷ ⑸。但本实验的工作未见报道。
本实验由齐齐哈尔大学化学化工学院的张晓慧、申书昌老师们,通过用微量进样器自制SPME装置,实现制备方法简单、价格低廉、有利涂层性能的研究。实验中,他们以苯乙烯和丙烯酸乙酯为单体,通过反应制备聚合物,并将此聚合物与气相色谱固定液(SE-30)混合制成复合SPME涂层。这种复合涂层热稳定性好,与纤维的结合力强,不易脱落。本文使用该复合涂层,采用HS-SPME-GC联用技术测试了水中氯代芳烃化合物,取得了满意的结果。他们初步实验结果优于国外产品,值得参考。现全文介绍如下。
2 实验部分
2-1 仪器与试剂
GC -122 气相色谱仪,火焰离子化检测器(上海分析仪器总厂),CDMC-4A色谱数据处理机(上海计算技术研究所),聚合反应装置(自己组装),Spectrum One傅里叶变换红外光谱仪(美国PE公司),热分析仪(美国PE公司,Pyris Diamond TG/DTA),石英纤维(100μm,武汉邮科所),5μ1微量注射器,不锈钢毛细管(内径0.47mm, 0.72 mm,赤山制管厂),磁力热搅拌器,千分尺,100m1具塞顶空瓶;
苯乙烯,丙烯酸乙酯,过氧化苯甲酰,甲醇,正己烷,浓硫酸,氢氧化钠,一氯苯,对二氯苯,1, 2, 4三氯苯,氯化钠,甲苯,三氯甲烷(以上试剂均为分析纯),色谱固定液(SE-30), OP-10乳化剂,重蒸馏水,水样(1.齐齐哈尔市劳动湖水,2.嫩江水,3.自来水)
2-2 聚合物的合成
2.2.1 单体和引发剂的精制
苯乙烯和丙烯酸乙酯分别用50g/L NaOH水溶液洗至中性,干燥后减压蒸馏,0.005MPa下收集60℃馏分,备用。引发剂(过氧化苯甲酰)用氯仿溶解后,再用甲醇使其析出,过滤、洗涤、真空干燥,备用。
2.2.2 苯乙烯一丙烯酸乙酯聚合反应
将一定量溶剂、引发剂(按引发剂g:单体kg = 3:1)及1/4单体混合物(苯乙烯kg :丙烯酸乙酯kg = 3.1:1) 同加人反应瓶中,安装好回流冷凝器,通氮气保护,在不断搅拌下控制反应温度开始反应。余下的3/4单体混合物倒人滴液漏斗中,控制速度,使其在1h滴完,反应一定时间后,得到的聚合物在60℃条件下减压到0.005Mpa,蒸出溶剂和剩余单体。反应瓶中聚合物用正己烷溶解,再用甲醇使其沉淀析出,弃去正己烷和甲醇,将胶状物干燥,备用。
2-3 SPME装置的研制
2.3.1 萃取头的制备
取 5cm 石英纤维浸泡在浓硫酸中,除去石英纤维表面的树脂。将聚苯乙烯丙烯酸乙酯与SE-30按质量比3:1混合于烧杯中,用氯仿和甲苯混合溶剂溶解,调整到一定的粘度后,转移至带塞小瓶中。将已处理好的石英纤维通过不锈钢小孔伸人小瓶,将粘稠液体涂渍到纤维表面上,经多次涂渍达到所需厚度及长度,在红外灯下除去溶剂。用特种粘合剂将涂制好复合涂层的石英纤维与细不锈钢管牢固结合,制成萃取头。
2.3.2 SPME装置的组装
将微量进样器内芯取出,去掉进人汽化室部分的不锈钢外套管,再将玻璃内不锈钢套管取出,在3.5μL刻度处断开玻璃管,后部分弃掉。将内径为0.72mm的不锈钢套管插人玻璃管内,并用粘合剂固定好,不锈钢管外露4cm,把带有萃取头的不锈钢内管穿人套管中,安装好紧固密封螺帽,防止漏气。玻璃内不锈钢套管的外露部分便是石英纤维进样保护套。
2-4 萃取实验
2.4.1 标准溶液配制
取0.4g一氯苯、0.4g对二氯苯、0.4g三氯苯,于1000m1容量瓶中,加人20g乳化剂(OP-10),用煮过的蒸馏水定容至刻度。溶液中一氯苯,二氯苯,三氯苯浓度均为400mg/L,此储备液至于阴暗处保存备用
2.4.2 色谱条件
色谱柱 2m×2mm ID.不锈钢柱,内填充 8% SE-30 202酸性红色担体(60-80目),柱温为140℃,检测器温度为180℃ ,汽化室温度为180℃;灵敏度为1010;载气(N2)为30ml/min,氢气为30ml/min,空气为300ml/min。
2.4.3 顶空萃取及色谱分析
2.4.3.1 工作曲线的制作
分别取lml、2 ml、4 ml、6 ml、8 ml、10ml标准溶液于六个l00ml顶空瓶中,加水至40ml,再加人20g氯化钠,将顶空瓶密封好,置于恒温水浴中,温度到达50℃,将萃取头穿过密封胶塞,置于瓶内进行顶空萃取,在磁力搅拌下萃取30min后,取出萃取头立即在气相色谱中解吸分析,根据色谱峰高和顶空瓶中各组分浓度分别作标准曲线。
2.4.3.2 样品分析
取三种样品各40ml于顶空瓶中,加人20g氯化钠,以下操作同标准曲线制作。根据色谱峰高定量,色谱分析见图-1。
3 结果与讨论
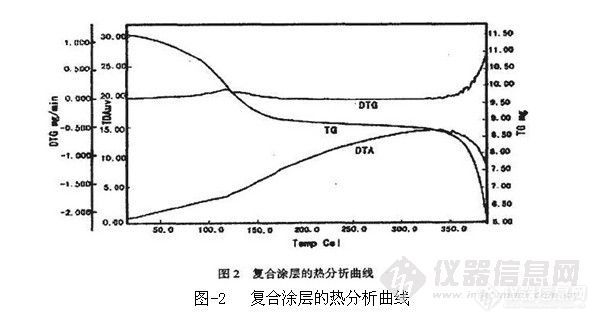
3.1 涂层的热稳定性
将涂制好聚苯乙烯丙烯酸乙酯涂层、SE-30涂层、聚苯乙烯丙烯酸乙酯与SE-30复合涂层的石英纤维分别倒挂在电热鼓风干燥器中,升温观察,结果发现,只涂有聚苯乙烯丙烯酸乙酯的涂层在90℃出现聚滴;SE-30涂层在160℃时出现聚滴,且同时出现脱落;聚苯乙烯丙烯酸乙酯与SE-30复合涂层,在300℃时出现聚滴。将聚苯乙烯丙烯酸乙酯与SE-30质量比为1: 1、2:1、 3:1 三种复合涂层进行比较,发现质量比为3: 1的复合涂层易于涂渍、热稳定性最好、不易脱落。复合涂层的热分析曲线见图-2。图-2表明了样品在320℃时才出现分解现象。因此本复合涂层最高使用温度可达290℃。将此萃取头插人290℃色谱气化室内,使用空柱(0.6m、柱温290℃)观察基线,基线平直。