-
+关注
私聊
-

w200761140
第101楼2011/07/20
(77讲)最后一讲
高效液相色谱一串联质谱法同时测定红小豆残留的6种咪唑啉酮类除草剂
摘 要: 建立了同时测定红小豆中6种咪唑啉酮类除草剂残留的高效液相色谱-串联质谱分析方法。样品经0.1mol/L NH4HCO3(pH 5)一甲醇(体积比为70:30)溶液提取,二氯甲烷液-液萃取和凝胶渗透色谱净化后,采用InertsilODS-3色谱柱(2.1 mm×150 mm,5μm)分离,以甲醇-0.1%乙酸为流动相梯度洗脱,离子阱质谱在选择离子模式下测定。咪唑啉酮类除草剂在10—200μg/L(灭草喹在5—100 μg/L)内线性关系良好,相关系数为0.9987—0.9997;方法的检出限为0.2—0.5μg/kg;在红小豆中3个加标水平的平均加标回收率为81.6%一99.4%,相对标准偏差为3.1%一7.8%。该方法简便、灵敏度高、精密度好,适用于红小豆中多种咪唑啉酮类除草剂残留的测定。
作者介绍: 本文由李成1, 锁然1, 王凤池2, 马宏颖1(1河北农业大学食品科技学院; 2. 河北省出入境检验检疫局)等研究人员共同完成并发表在《色谱》第二十六卷第六期上的论文,主要联系人是李成,硕士研究生,主要从事食品分析研究.Tel:(0312)7528195,E-mail: 1ccpopl@yahoo.com
前 言
咪唑啉酮类除草剂是一种高效、广谱、高选择性除草剂,问世至今这种除草剂已经开发有6种商品化品种,包括咪唑烟酸(imazapyr)、甲氧咪草烟(imazamox)、甲基咪草烟(imazapic)、咪唑乙烟酸 (imazethapyr)、灭草喹(imazaquin)、咪草酸酯(imazamethabenz.methyl)[1]。这类除草剂在我国主要用于防除豆类、花生、玉米等农作物的田间杂草。虽然其使用量小,但多属于长效除草剂,在土壤中的微量残留对后茬作物造成药害[2]。近年来,各国对此类除草剂做出了严格的限量要求。2005年7月,加拿大经过对相关数据审核,确定大豆内的咪唑乙烟酸的最高残留限量为0.1 mg/kg。2006年2月,美国出台了“G/SPS/N/U&V1229最终法规”,该法规要求稻米中咪唑乙烟酸总残留限量不得高于0.3 mg/kg。2006年5月,日本正式实施的“食品残留农业化学品肯定列表制度”对不同食品中咪唑啉酮类除草剂的限量范围为0.01—0.5 mg/kg。这些新的限量标准的实施,给我国的农产品出口设置了新的技术壁垒,因此建立一种食品中咪唑啉酮类农药残留的简便、准确、高灵敏度的检测方法是迫切而有重要意义的。
目前国内外检测咪唑啉酮类除草剂的方法主要为气相色谱一质谱联用法(GC-MS)[3]、毛细管电泳-质谱联用法(CE—MS)[4]、高效液相色谱法[5-10]、高效液相色谱-质谱联用法(LC-MS)[11-14]等。分析的对象主要是土壤、水等环境样品中一种或几种农药成分的检测,在食品类基质中6种除草剂的同时检测方法未见报道。本文采用液-液萃取和凝胶渗透色谱净化样品,建立了红小豆中6种咪唑啉酮除草剂痕量残留的高效液相色谱一串联质谱检测方法。该方法样品净化效果好、灵敏度高,可满足红小豆中多种咪唑啉酮类除草剂残留检验工作的需要。
1 实验部分
1.1 仪器与试剂
Agilent 1100高效液相色谱仪,配有电喷雾离子源(ESI)、离子阱质谱系统(美国Agilent公司);液相色谱柱:Inertsil ODS-3(150 mm×2.1 mm,5μm)(日本Dikma公司)。Accuprerp凝胶色谱(GPC)仪、凝胶渗透色谱柱(22 g S—X3 Bio-Beads填料,粒度200—400目)(美国J2 Scientific公司);二氯甲烷、甲醇、乙酸乙酯、环己烷均为色谱纯(美国Tedia公司);水为超纯水(经0.45μm滤膜过滤)。
咪唑啉酮类标准品:咪唑烟酸、甲氧眯草烟、甲基咪草烟、咪唑乙烟酸、灭草喹、咪草酸酯(纯度≥98.0%,德国Dr.Ehrenstorfer公司)。
标准溶液的配制:分别准确称取标准品10 mg(精确至0.1 mg),用乙腈溶解并定容于100 mL容量瓶中,配制成100 mg/L的标准储备液,冷冻避光保存于4℃冰箱中。根据需要,取适量的标准储备液用乙腈稀释至所需浓度的标准溶液。
1.2 实验方法
1.2.1 样品前处理方法
样品经粉碎后过40目筛。称取5.00 g(准确至0.01 g)于150 mL三角瓶中,加入0.5 g硅藻土、50mL 0.1 mol/L NH4HCO3(pH 5)一甲醇(体积比为70:30)溶液,在振荡器上振荡提取30 min。减压抽滤提取液,用10 mL提取剂洗涤三角瓶及滤渣,合并滤液于150 mL浓缩瓶中。在45℃水浴中将提取液浓缩至约20 mL,加入20 mL 150 g/L氯化钠溶液,用1 mol/L HCl调pH值为2,转移至分液漏斗中,用40 mL二氯甲烷分两次洗涤浓缩瓶,合并至同一分液漏斗中,振摇萃取2 min,静置分层。二氯甲烷层经5 g无水硫酸钠过滤至浓缩瓶中,用40mL二氯甲烷重复萃取一次,合并二氯甲烷于同一浓缩瓶中,在45℃水浴中蒸至近干。残渣用GPC流动相溶解转移至GPC专用小管中,定容至10mL,于5 000 r/min下离心5 min。上清液经GPC净化,弃去淋洗液,将收集液旋转蒸发至约2 mL,用氮气吹干,用液相色谱流动相定容至1 mL,过0.45μm有机系滤膜,滤液供LC-MS/MS测定。
1.2.2 GPC净化条件
流动相:乙酸乙酯-环己烷(体积比为1:1);流量:5 mL/min;进样量:5 mL;检测波长:254min;净化程序:0→8 min,淋洗,弃去淋洗液;8→14 min,收集洗出液。
1.2.3 LC—MS/MS分析条件
液相色谱条件:流动相为甲醇(A相)和0.1%乙酸(B相),流速0.3 mL/min;梯度洗脱程序:O→6 min,40%A→43%A;6→7 min,43%A;7→l 1 min,43%A→70%A;11→19 min,70%A。柱温30℃。进样体积15μL。
MS/MS条件:电喷雾电离(ESI),正离子化模式;毛细管电压4 000 V;雾化气压力314 kPa;干燥气流速9 L/min;干燥气温度350℃。
2 结果与讨论
2.1 提取剂和萃取条件的选择
咪唑啉酮类除草剂有较强的极性,提取效果受pH值的影响较大。分别用乙腈、甲醇、乙酸乙酯、丙酮-水、甲醇-水作提取剂进行样品添加试验,添加回收率表明上述提取剂均不能实现对6种除草剂的同时提取。选用不同比例的0.1 mol/L NH4HCO3(pH 5)·甲醇作为提取剂分别进行添加试验,结果表明:O.1 mol/L NH4HCO3(pH 5)-甲醇(体积比为70:30)作提取剂时的添加回收率均达到85%以上。用1 mol/L HCl分别调提取剂的pH为5、6、7、8以优化提取剂的pH值,
结果表明:pH值为5和6时,添加回收率均能达到85%以上;提取剂的pH为7时,6种除草剂的添加回收率明显降低;提取剂的pH为8时,除咪草酸酯的添加回收率为86%外,其他除草剂的添加回收率均低于20%。综合考虑提取剂的极性和提取剂的pH值对提取效果的影响,本文选用0.1 mol/L NH4HCO3(pH 5)-甲醇(体积比为70:30)作为提取剂。液-液萃取时浓缩后的提取液的pH值大于咪唑啉酮类除草剂的pK,值⋯,因此多数咪唑啉酮类除草剂以离子形式存在,用二氯甲烷直接萃取的效果不好。试验表明:液-液萃取时不调节pH值,咪唑烟酸回收率只有16.6%,其他5种除草剂的回收率也均低于50%。液-液萃取时用1 mol/L盐酸调节不同的pH值进行对比试验,结果表明:pH值为1.5~2.5时6种除草剂回收率能达到90%以上。故本文液-液萃取时将pH值调至2。
2.2 GPC净化条件的确定
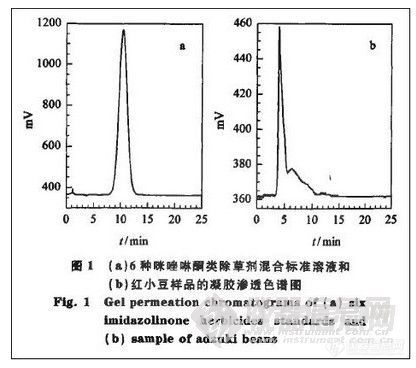
由于红小豆中含有蛋白质、脂类、色素等大分子物质,故采用GPC方法净化。配制合适浓度的6种除草剂的混合标准液,用GPC流动相定容,在检测波长254 nm下过GPC柱得到的色谱图见图1一a,可见6种除草剂于8~13 min出峰。在同样条件下做样品空白试验,其色谱图(见图1-b)中只在4 min处有最大吸收峰杂质,表明GPC净化可以去除大部分杂质。用样品加标试验验证GPC条件,将收集洗出液的时间设为8—13 min,测得的回收率除咪草酸酯和甲基咪草烟外,其他4种除草剂均低于80%,认为可能是基质对分析物有影响。做5组样品的加标试验,分别设收集时间为7—13 min,7.5~13min,8—13 min,8.5—13 min,9—13 min 5个收集时间,结果表明:在7—13 min,7.5~13 min和8—13 min收集洗出液,6种除草剂的回收率基本相同,均在70%~90%之间;在8.5~13 min和9—13 min收集,除草剂的回收率明显降低。因此推断分析物受基质影响出峰时间后移,将起始收集时间点设为8 min。再做4组样品的加标试验,分别于8—13.5min,8~14 min,8~14.5 min和8—15 min 4个不同的收集时间段收集洗出液,试验结果表明:于8—13.5 min收集,6种除草剂的回收率达到85%以上;分别于8~14 min,8~14.5 min和8~15 min收集,6种除草剂的回收率基本相同,均在95%以上。通过两组样品的空白加标试验,最终确定GPC洗出液的收集时间为8—14 min。
2.3 色谱条件的选择
6种咪唑啉酮除草剂的极性较大,以有机相.水相作流动相时分析物出峰早,分离度差。考虑pH值对6种除草剂结构的影响,本文分别以乙腈和0.1%乙酸、甲醇和0.1%乙酸作流动相对色谱条件进行优化。试验表明:以乙腈和0.1%乙酸作流动相梯度洗脱难以使甲基咪草烟和甲氧咪草烟达到基线分离,而以甲醇和0.1%乙酸作流动相梯度洗脱可以使6种除草剂达到基线分离,在19 min内6种除草剂全部出峰。
2.4 质谱条件的选择
6种咪唑啉酮类除草剂均为极性化合物,根据分子结构的特征,试验选择了ESI(+)作为电离模式。以蠕动泵10μL/min的流速,分别注射0.1mg/L的6种除草剂标准溶液进ESI电离源,在正离子扫描方式下分别对分析物进行一级质谱分析,得到分子离子峰,然后对各准分子离子峰进行二级质谱分析(子离子扫描),得到碎片离子信息,确定定性和定量子离子,并优化仪器的各个参数使其响应值最大。6种咪唑啉酮类除草剂质谱参数和保留时间见表1。
2.5 线性关系、回收率、精密度、检出限及定量限
大量试验发现在LC-MS/MS法检测咪唑啉酮类除草剂时存在基质效应,在相同浓度下用基质配制的标准溶液的响应值比用纯试剂配制的标准溶液的响应值要低。因此,本文选用基质提取物配制一系列不同浓度的混合标准溶液进行测定,以各组分的峰面积Y 对质量浓度X(μg/L)做线性回归。结果表明:6种咪唑啉酮类除草剂在10—200μg/L(灭草喹在5-100μg/L)范围内线性良好,相关系数均在0.9987—0.9997之间,可以满足定量分析的要求。在红小豆空白样品中添加3个水平的6种咪唑啉酮类除草剂混合标准溶液,每个水平高6组平行试验,按照方法进行回收率测定,计算添加回收率及相对标准偏差(RSD)。结果表明:咪唑啉酮类除草剂的回收率在81.6%~99.4%之间,6次测定结果的RSD为3.1%一7.8%,结果见表2。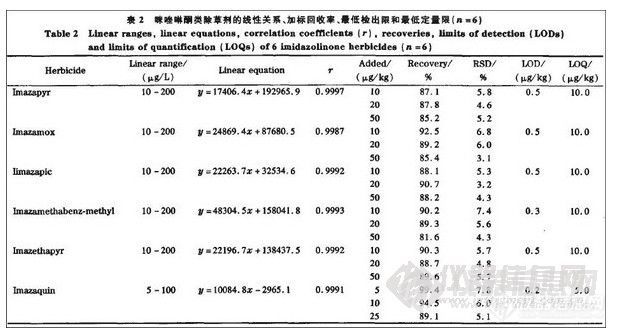
该结果表明方法的准确度和精密度均符合农药残留分析的要求。依照上述建立的方法测定基质空白的噪声,以3倍信噪比峰高对应的含量为检出限(LOD),其范围为0.2-0.5μg/kg;以大于10倍信噪比峰高对应的含量为最低定量限(LOQ),其范围为5.0—10.0μg/kg,相应的色谱图见图2。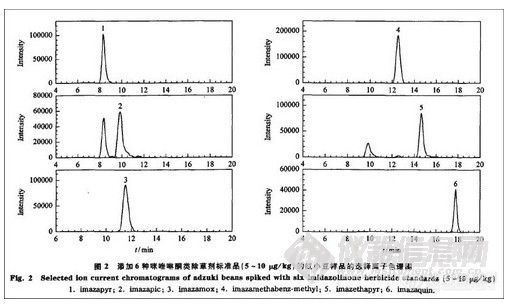
2.6 实际样品的测定
用本文建立的方法分别测定了12份红小豆样品(采自当地农贸市场)中6种咪唑啉酮类除草剂残留,结果均未检出该类除草剂。
3 结论
根据咪唑啉酮类除草剂的结构特性,在提取和净化过程中选择了合适pH值的提取剂提取目标组分,使回收率达到了理想的效果;对于红小豆中的蛋白质、脂类、色素等大分子物质,采用凝胶渗透色谱净化的方法去除了大部分干扰物质,取得了理想的净化效果。本文建立的凝胶色谱净化结合高效液相色谱.串联质谱对红小豆中残留的6种咪唑啉酮类除草剂的分析方法灵敏度高,选择性强,能够满足农药残留检测要求。
