-
+关注
私聊
-

yhl-87_
第81楼2009/11/06
农药残留检验方法系列讲座(61)(下)固相萃取-高效液相色谱法测定苹果中 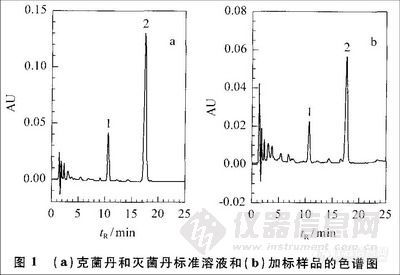
2.3 克菌丹和灭菌丹测定波长的选择
克菌丹和灭菌丹的紫外吸收光谱见图2。克菌丹的最大吸收波长为207 nm,灭菌丹的最大吸收波长为222 nm。克菌丹在254 nm处没有吸收峰,因此SN0654-1997方法选用254 nm测定克菌丹的灵敏度很低。用HPLC分析克菌丹和灭菌丹时、流动相中的甲醇、乙腈的吸收峰在195 nm 左右,乙酸-乙酸钠缓冲溶液的主吸收峰在200nm之前。为了能够同时测定克菌丹和灭菌丹,选择检测波长为210 nm。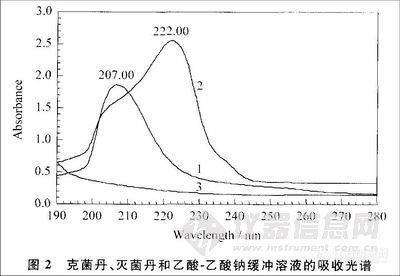
2.4 流动相pH值的影响
实验证明克菌丹和灭菌丹在pH 值为0.5~5.0时很稳定。当pH值高于5.3后,克菌丹和灭菌丹将发生分解。由于Eclipse XDB-C18柱使用的最佳pH值范围为3~8,故本文研究了流动相pH值对克菌丹和灭菌丹分离的影响 结果表明pH值在3.55~4.87范围内,对克菌丹和灭菌丹分离效果影响很小,并且峰面积的变化也很小,故选用流动相pH为3.80。
2.5 检出限、线性范围及线性关系
在最佳测定条件下,克菌丹和灭菌丹的保留时间及重现性、线性范围、相关系数、线性方程及最小检出限(以3倍信噪比计算)见表1。在所测定的含量范围内,被测物质的含量(X)与峰面积(Y)之间的相关系数大于0.9999.保留时间的相对标准偏差(RSD)≤0.60%,检出限为0.20~0.27 mg/kg.
2.6 混合吸附剂固相萃取柱与其他固相萃取柱的比较
目前测定水果中克菌丹和灭菌丹时采用Florisil固相萃取柱和氨基固相萃取柱净化提取样品。本文考察了国产硅镁吸附剂和硅胶混合吸附剂对苹果的净化提取效果,并与Florisil柱和氨基柱的净化提取效果进行比较,结果见表2。表2的数据表明,氨基柱对克菌丹和灭菌丹的吸附比较强,目标组分不能完全被洗脱下来,导致平均回收率只有50%左右,且重现性较差。Florisil柱和国产硅镁吸附剂与硅胶混合固相萃取柱对克菌丹和灭菌丹的净化效果较好。用Florisil柱净化时。克菌丹和灭菌丹的平均回收率为l07%~111%,相对标准偏差为4.6%~6.9%。用自制的硅镁吸附剂与硅胶吸附剂混合固相萃取柱净化,克菌丹的7次平均加标回收率为104%,相对标准偏差为5.2%;灭菌丹为99.0%,相对标准偏差为7.5%;但自制的混合吸附剂固相萃取柱比商品Florisil柱的成本低。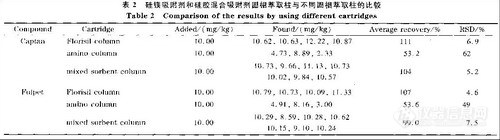
2.7 洗脱溶剂对测定克菌丹和灭菌丹的影晌
洗脱溶剂的选择对农药分析中的样品净化效果有较大影响。本文比较了25%丙酮的正己烷、75%丙酮的环己烷和二氯甲烷-正己烷-乙腈(体积比为49:50:1)混合溶液作为洗脱溶剂净化苹果中克菌丹和灭菌丹的效果(见表3) 3组溶剂均能够完全洗脱克菌丹和灭菌丹,但用75%丙酮的环己烷作洗脱溶剂时,克菌丹和灭菌丹的回收率大于120%;用25%丙酮的正己烷和二氯甲烷一正己烷-乙腈作洗脱溶剂时,克菌丹和灭菌丹的回收率均符合要求,而二氯甲烷-正己烷一乙腈的效果最佳,两种组分的回收率均为102%,7次测定的RSD小于5.0%。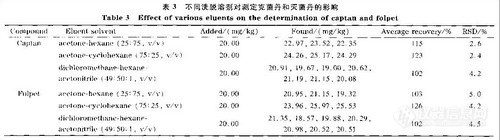
2.8 实际样品的测定
按照实验方法,确称取25.00 g苹果匀浆样品20份, 其中5份测定样品中克菌丹和灭菌丹的实际含量,另外15份样品加入不同浓度水平的克菌丹和灭菌丹的混合标准溶液,测定其加标回收率,以考察方法的准确性和可靠性,结果见表4。
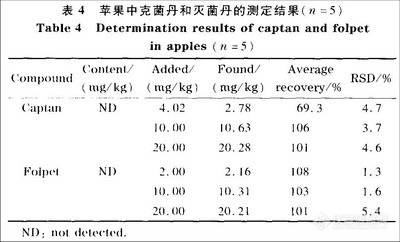
表4结果表明,克菌丹的平均加标回收率为69.3%~106%,RSD为3.7%~4.7%,灭菌丹的平均加标回收率为101%~108%,RSD为1.3%~5.4%。
参考文献
-
+关注
私聊
-
yhl-87_
第82楼2009/11/06
农药残留检验方法系列讲座(62)三氟氯氰菊酯农药中微量杂质的液相色谱-质谱法鉴定
摘 要: 用高效液相色谱- 质谱联用技术研究了三氟氯氰菊酯农药的杂质及含量, 并用源内碰撞诱导解离(CID) 技术鉴别了所存在的微量杂质。方法实用性强, 色谱分离效果好, 可用于该农药各成分的定性、定量分析
本文由上海市计量测试技术研究院的查月珍研究人员完成,摘自“分析测试学报”第20卷第一期。
前 言
三氟氯氰菊酯属于拟除虫菊酯类农药, 它的特点是高效、毒性中等、残留量低, 因而是国内外应用最广的农药种类之一[ 1 , 2 ]。随着环境科学的发展, 人们对环境保护的意识越来越强。评价一种农药的好坏除了它的有效性外, 它的安全性还应包括产品中带来的微量杂质对环境的污染作用, 因此, 国外对农药除了有效成分有一定的要求外, 对杂质的种类和含量也有一定的要求。随着液相色谱- 质谱(LC- MS) 联用技术的不断完善, LC- MS 在农药分析中发挥着日趋重要的作用, 在很大程度上替代了传统的气相色谱- 质谱联用技术, 但通过液相色谱- 质谱联用法来测定三氟氯氰菊酯农药中的杂质和含量还未见报道。本文用高效液相
色谱- 质谱联用技术研究了三氟氯氰菊酯农药的杂质及含量, 并用源内碰撞诱导解离技术(CID) 鉴别了所存在的杂质。方法简便可靠, 色谱分离效果好, 用普通的C18 反相柱使三氟氯氰菊酯中的异构体也得到了良好的分离, 可用于该农药各成分的定性、定量分析。
实 验
1 实验部分
1. 1 仪器与试剂
Agilent HP1100/ MSD 高效液相色谱- 质谱联用仪, 包括自动进样器, 柱温箱和二极管阵列检测器(DAD) 。甲醇为HPLC级, 水为超纯水; 三氟氯氰菊酯样品由江苏省激素研究所提供, 用甲醇溶解样品配制成2 g/ L的分析液。
1. 2 高效液相色谱- 质谱联用条件
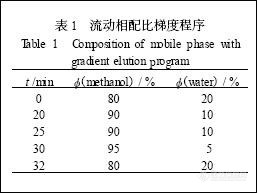
色谱条件: 色谱柱Inertsil ODS - 3 柱(2. 1 mm×150 mm, 5μm) ;流动相甲醇+ 水梯度洗脱,梯度程序见表1 ; 流速0. 2 mL/ min , 柱温30 ℃, 进样量3μL ; DAD 检测波长278 nm。
质谱条件: 电喷雾正负离子检测, 源内CID 电压分别为50 V、100 V、120 V、140 V, 质量扫描范围m/ z 70~630 , 步长0. 10 , 扫描速度1. 01 s (全程) , 峰宽0. 1 min。
2 结果与讨论
2. 1 结果
图1、图2 分别为三氟氯氰菊酯的色谱图和总离子流图, 可见质谱响应良好。从它们的质谱图可以清楚地看到分子离子峰和碎片峰, 其结果列于表2。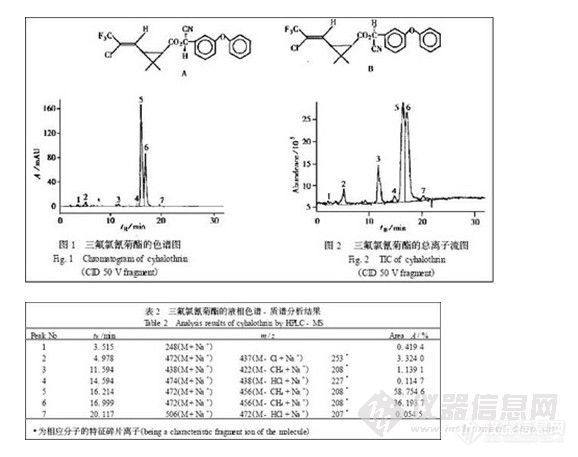
2. 2 讨论
提供分析样品的有效成分是三氟氯氰菊酯的两个非对映异构体组成的混合物。它们是cis- Z- (1 R) (结构式A) 和cis- Z- (1 S) (结构式B) [ 3 ]
在谱图中的5 号、6 号峰为这一非对映异构体, 分离良好, 质谱信号响应强。为了能有效地鉴别杂质成分, 实验中分别用了50 V、100 V、120 V、140 V进行测试, 表2 中的碎片离子是在不同的CID 电压下得到的。实验中发现, 样品结构中虽然有N 原子, 但H+ 离子的加入不能增加质谱的灵敏度, 相反使质谱信号本底有明显增加, 可能是N原子受到空间位阻的屏蔽作用; 相反, Na + 离子很容易与此化合物缔合, M+ Na + 峰非常强, 使含氯的质谱信号特征清楚。从谱图中保留时间为4. 978 min 的2 号峰的相对分子质量和其碎片离子的质量表明: 产品中还有微量的其它异构体杂质; 3 号峰不含氯, 4 号峰双键被打开, 7 号峰含二个氯; 1 号峰在正离子检测中质谱信号非常弱, 很难判别m/ z 为248 的相对分子质量, 故又采用负离子扫描, 得到了m/ z 为260 , 且带有一个Cl 的特征质谱信号, 因此, 可以肯定1 号峰的相对分子质量为225 , 其正离子质谱为M+Na + , 负离子质谱为M+ Cl - 。遗憾的是随着CID 电压的增加, 1 号峰的质谱信号迅速减弱, 故未能得到结构信息。作者曾经试图用乙腈作流动相, 发现乙腈的存在使该化合物的质谱信号大大减弱, 并且不能分离该化合物的异构体。
-
+关注
私聊
-
yhl-87_
第83楼2009/11/06
农药残留检验方法系列讲座(63)蔬菜中有机氯和拟除虫菊酯农药残留量的测定
摘 要:目的建立了蔬菜中8种有机氯农药和4种拟除虫菊酯农药残留气相色谱分析方法。方法样品用丙酮浸泡过夜。再用石油醚进行液液分配,提取液用弗罗里硅土柱净化,采用SE一54弹性石英毛细管柱分离,用GC—ECD同时检测。结果高、中、低3个水平添加时的回收率为87.5%~104.7%。相对标准偏差为2.2%~5.8%。该方法的检出限为:有机氯农药质量分数为5.0×10-8%~7.0×10-7%,拟除虫菊酯质量分数为5.0×10 -7%~8.0×10-7%。结论可用于蔬菜中农药残留量测定。
本文由沈阳药科大学药学院的邹继红,宋欣鑫,王静和赵春杰与东北制药总厂的季光辉合作完成,现全文介绍如下。第一作者邹继红硕士联系方式024—23986299;最后一位赵春杰教授主要从事农药残留分析测定,联系方式zcjjljj@sina.com
前 言
农产品(食品)的安全性问题是当今全球关注的焦点之一。而农产品(食品)中的农药残留又是其中一个非常重要和突出的问题。有机氯农药是一类广谱杀虫剂,属于神经毒物,可致癌。该类农药性质稳定,不易分解,能在水域、土壤和生物体内长期贮存。经植物富集后会导致严重污染。拟除虫菊酯类农药大多为广谱性杀虫剂,已用于粮食、蔬菜、水果等农作物。虽然此类农药易被人体内氧化酶降解,无蓄积作用,一般认为是低毒性农药,但若使用不当或食用其污染食品,亦会引起中毒。已有的有机氯农药残留检测方法中多采用磺化法[l~5]净化,而本文采用弗罗里硅土柱净化使样品更纯净。菊酯类农药残留检测虽然已有报导[6~8],但同时测定2类农药残留的方法很少,今用毛细管气相色谱法对两类农药残留进行测定。取得了较好的结果。
实 验
1仪器与材料
Shimadzu GC一17A气相色谱仪、Shimadzu63Ni电子捕获检测器、CIASS_GClO色谱工作站(日本岛津公司),SE一54弹性石英毛细管柱 (30 m×0.32 mm×1.0μm,大连中汇达公司)。农药标准品:六六六(BHC),包括α一BHC、β—BHC、γ—BHC和δ一BHC
4种异构体、氯菊酯(pemethrin)、S一氰戊菊酯(fenvalerate)、溴氰菊酯(deltamethrin)、甲氰菊酯(fenpropathrin) (纯度质量分数>99%,均购自国家农药标准品中心),滴滴涕(DDT),包括pp’DDE 、 pp’DDD、 pp’DDT 和op’DDT 4种同系物,纯度质量分数>98%,均购自美国Sigma公司。甲醇为色谱纯,水为重蒸馏水,其他试剂均为分析纯。
2方法
2.1色谱条件
采用毛细管气相色谱法。色谱条件为:SE一54弹性石英毛细管柱(30. m×0.32 mm×1.0μm),进样口温度:260℃,检测器温度:320℃,柱升温程序:初始温度140℃,以
10℃·min-1升至270℃保持5 min。以20℃·min-1升至290℃保持15 min,载气:高纯
氮;流量:50 mL·min-1进样量:1μL。
2.2标准曲线的绘制
精密称取α一BHC、β—BHC、γ—BHC和δ一BHC 4种异构体、pp’DDE 、 pp’DDD、 pp’DDT 和op’DDT 4种同系物、甲氰菊酯、氯菊酯、S一氰戊菊酯、溴氰菊酯各1 mg,分别于25 mL量瓶中,加正己烷溶解,定容。
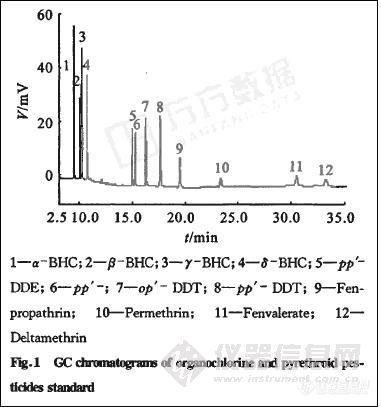
分别精密吸取12种农药适量置于100 mL量瓶中。加正己烷稀释至刻度,得有机氯和菊酯类农药的混合对照品贮备液,精密吸取上述混合溶液用正己烷逐步稀释成6个质量浓度梯度的混合对照品液,按“2.1”条色谱条件,分别吸取上述6份标准溶液各1μL进样,每份进样5次。以峰面积为纵坐标,农药的质量浓度为横坐标,计算其回归方程,见表1。结果表明,在10~4 000 mg·L-1呈良好线性。色谱结果如图1所示。
2.3样品制备
称取50 g充分混匀的样品。加80 mL丙酮浸泡过夜,于多功能搅拌机中搅碎,经抽滤,滤液转入500 mL分液漏斗中,加150 mL质量分数为2%的Na2S04溶液和50 mL石油醚,振摇萃取,无机相再分别用30 mL石油醚萃取2次。合并石油醚相于250 mL磨口瓶中,于60~70℃水浴上旋转蒸发至约2 mL,通过石油醚预淋的层析柱(层析柱由下至上:少许玻璃棉+2 cm无水硫酸钠+4 cm弗罗里硅土+2 cm无水硫酸钠),再用75 mL石油醚分多次冲洗柱子,滤液收集于250 mL磨口烧瓶中,旋转蒸发至约2 mL,再转入10 mL容量瓶,用石油醚定容。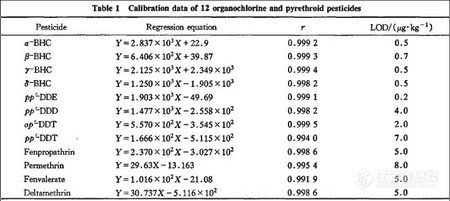
3结果
3.1出峰顺序
配制一系列低质量浓度标准品溶液进样,在“2.1”色谱条件下出峰顺序为:α一BHC、β—BHC、γ—BHC、 PCNB、δ一BHC 、pp’-DDE 、 pp’-DDD、 op’DDT、 pp’DDT、甲氰菊酯、氯菊酯、S一氰戊菊酯、溴氰菊酯。
3.2精密度试验
3.2.1 色谱系统精密度试验
吸取40 mg·L-1对照品溶液1μL,重复进样’5次,按色谱条件下操作得α一BHC、β—BHC、γ—BHC、 PCNB、δ一BHC 、pp’-DDE 、 pp’-DDD、 op’DDT、 pp’DDT、甲氰菊酯、氯菊酯、S一氰戊菊酯、溴氰菊酯的RSD(%)分别为2.6、3.2、3.5、2.9、4.4、3.8、5.7、3.6、4.4、4.8、3.5、3.0。
3.2.2方法精密度试验
准确称取同一样品5份,按“2.3”条方法操作,按“2.1”条方法测定,各农药的RSD(%),分别为2.4、3.3、4.7、2.8、3.6、3.5、4.1、4.5、3.7、3.8、4.2、4.7。
3.3回收率试验
精密称取同一样品9份。分3个水平添加混合标准品溶液,每水平重复3次,混匀。按“2.3”方法提取、净化、测定并计算回收率,结果见表2。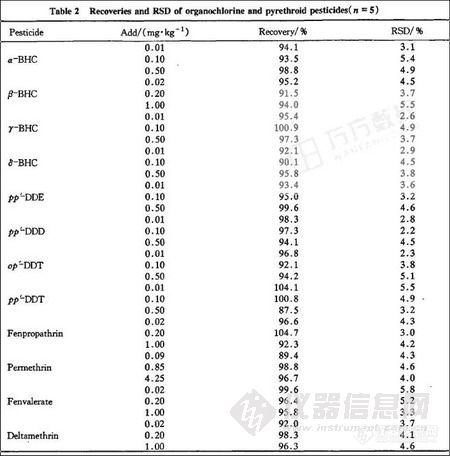
3.4样品的测定
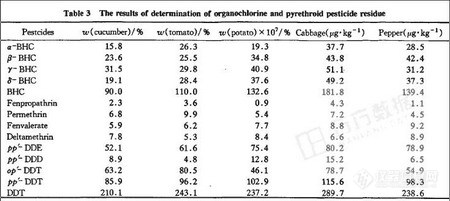
准确称取蔬菜样品,按“2.3”条测定,每份样品重复2次,每次进样1 μL,用40 μg·L-1混合标准溶液做随行标准,用外标法定量。样品测定图谱见图2,结果见表3。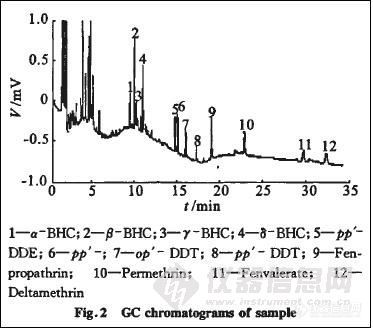
4讨论
a.曾采用丙酮浸泡1 h,超声提取30 min来处理样品,结果提取不完全,所以改为丙酮浸泡过夜。
b.根据以往经验和文献报道,在分析植物试样中有机氯和拟除虫菊酯类农药的前处理净化方法中。常用的吸附剂有弗罗里硅土、酸性/中性氧化铝、硅胶、活性炭等,而弗罗里硅土比较适合有机氯和拟除虫菊酯两类农药的净化。故选用弗罗里硅土柱净化。
C.根据测定有机氯和拟除虫菊酯农药时的称样量、定容体积、标准物质的响应值、ECD的灵敏度、空白试样的噪声水平等因素来确定每个农药的检测限。
d.从表3中可以看出,由于有机氯农药的稳定性好,半衰期长,虽然在我国禁用已有近20年,但在蔬菜瓜果中仍有一定量的残留,而拟除虫菊酯类农药在蔬菜水果种植过程中未发现有大量使用。

-
+关注
私聊
-
yhl-87_
第84楼2009/11/06
农药残留检验方法系列讲座(64)气相色谱-质谱法分析蜂蜜中的多种农药残留
摘 要:开展了蜂蜜中23种农药残留的气相色谱-电子轰击离子源质谱GC-EI/MS分析方法的研究,并对其中3种农药的EI/MS碎片离子的断裂机理与结构进行了初步解析。探讨了蜂蜜试样前处理条件的优化与选择。将蜂蜜试样用乙酸乙酯提取剂超声提取、Florisil硅藻土色谱柱净化和正己烷-乙酸乙酯( 体积比为7:3)混合洗脱剂洗脱后,以PCB103为内标物,采用选择离子监测(SIM)方式下的GC-EI/MS分析。当试样的加标浓度为50,100 和200μg/kg时,加标回收率为82%~120%,相对标准偏差小于11.0%。23种农药的检测限都小于10.0μg/kg,线性范围为10~500μg/kg,相关系数都大于0.995。此分析方法已成功地应用于蜂蜜中23种痕量农药残留的分析。
本文由 (1)厦门大学化学化工学院化学系现代分析科学教育部重点实验室和(2)龙岩学院化学与材料工程系的金珍1、林竹光1、陈美瑜1、 马玉1、谭君1、范玉兰1、翁嘉辰1、陈招斌1、涂逢樟1,2等共同完成。蜂蜜中的活性物质多,微量残留农药的分析比较困难,本文做了较全面的阐述值得参考,现全文介绍如下。
进一步的联系人林竹光副教授 0592-2184660 Email linzg@xmu.edu.cn 第一作者是当时的研究生
前 言
蜂蜜作为一种天然、营养和保健食品备受欢迎。20世纪90年代以来,国外对蜂蜜食品的质量、卫生和安全的要求越来越严格,对蜂蜜中农药残留的分析也有严格的技术标准。Jimenez等利用Florisil硅藻土净化、气相色谱(GC)的选择性检测器分析了蜂蜜中23种农药残留[1],并研究了固相微萃取法在蜂蜜中多种农药残留分析的应用[2]。Rissato 等[3]利用超临界流体萃取GC电子捕获检测器(ECD)分析了蜂蜜中有机氯、有机磷、氨基甲酸酯和拟除虫菊酯等农药的残留。Beatriz 等[4]利用固相萃取GC质谱(MS)分析了蜂蜜中51种农药残留。Albero等[5]利用基质固相分散-GC-ECD分析了蜂蜜中有机氯、有机磷和拟除虫菊酯类等15种农药残留。目前,我国关于蜂蜜中农药残留的标准分析方法大多是单类和单种,如氟胺氰菊酯农药残留的分析[6~7]。GC-MS 同时分析蜂蜜中多种农药残留的报道较少[8]。
本文以超声辅助提取,色谱柱净化,选择离子监测方式(SIM)下的气相色谱!电子轰击离子源质谱法(GC-EI/MS)和内标法同时分析蜂蜜中氨基甲酸酯、有机磷和拟除虫菊酯类等23种农药的残留量。
实 验
1. 实验部分
1.1仪器与试剂
仪器:shimadzu GC/MS-QP 2010气相色谱-质谱联用仪( 日本岛津公司);KQ 3200E超声波清洗器( 江苏昆山市超声仪器有限公司);DK-S22 型电热恒温水浴锅( 上海精宏实验设备有限公司);自制的氮吹浓缩装置。
试剂:甲醇、丙酮、正己烷和乙酸乙酯均为农残级试剂( 美国Tedia公司);无水Na2SO4 ( 分析纯),于650℃马弗炉中烘烤4h;Florisil硅藻土( 分析纯),100~200目,于650℃马弗炉中烘烤4h,使用前再于140℃烘箱中烘2h,加8%超纯水去活。
农药标准物质:速灭威、叶蝉散、仲丁威、呋喃丹、抗蚜威、西维因、乐果、二嗪磷、甲基对硫磷、杀螟松、马拉硫磷、毒死蜱、倍硫磷、对硫磷、稻丰散、乙硫磷、伏杀硫磷、联苯菊酯、甲氰菊酯、氯菊酯、氯氰菊酯、氰戊菊酯和溴氰菊酯( 中国农业部环境保护科研监测所);PCB103( 2,2’,4,5’,6-pentachlorobiphenyl)内标物( IS)( 美国Accu standard公司)。
内标物的配制:取1mL 35g/L PCB103内标物,将其溶于正己烷溶液中稀释成所需要的浓度。
1.2 蜂蜜试样的提取与净化
提取:将蜂蜜试样置于40℃恒温水浴中加热10min,准确称取1.00g试样于50mL具塞三角瓶中,加入1.00mL甲醇溶剂振荡摇匀。用10mL乙酸乙酯提取剂超声提取10.min,转移出上层有机提取液;残渣再用5mL乙酸乙酯提取剂超声提取10min。合并两次提取液,加适量无水Na2SO4除水后,氮吹浓缩至5mL。
净化:在20cm ×1.5cm I D玻璃色谱柱内填入适量的玻璃毛,再依次填入1cm高无水Na2SO4 2.0g Fkorisil 硅藻土和1cm高无水Na2SO4。先用10mL 正己烷预淋洗色谱柱,再将浓缩后的提取液转移至色谱柱内,然后用15mL正己烷:乙酸乙酯( 体积比为7:3)混合洗脱剂洗脱,洗脱液氮吹浓缩近干,加入1.00mL 10μg/L PCB103 内标物溶液溶解于带刻度的小测试瓶中,氮吹定容至1.00mL,供下一步进行仪器分析。
1.3GC-EI/MS分析条件
GC分析条件:DB-5ms毛细管柱(30m×0.25mm×0.25μm );载气为He( 纯度99.999% );柱头压58.8kpa;载气恒线速度36.8cm/s;不分流进样,进样体积1.00μL;进样口温度250℃;GC-MS接口温度250℃。色谱柱升温程序:60℃( 保持1.0min),以30℃/min的速率升至150℃,再以8℃/min的速率升至280℃( 保持10min)。
EI/MS 分析条件:电子能量70eV;灯丝电流60μA;检测器电压1.00kv;EI源温度200℃;溶剂延迟时间3.0min。
2. 结果与讨论
2.1 蜂蜜试样前处理条件的优化与选择
多种农药残留分析采用的提取剂较为特殊,相对于拟除虫菊酯类农药,有机磷和氨基甲酸酯类农药的极性较强,若仅以正己烷为提取剂,则提取效率偏低,因此必须选择二氯甲烷、乙酸乙酯和丙酮等极性较强的溶剂或它们的混合溶液为提取剂。
分别以正己烷:二氯甲烷( 体积比为1:1)、正己烷:丙酮( 体积比为1:1)、正己烷:乙酸乙酯( 体积比为1:1)和乙酸乙酯为提取剂做提取效率对比试验,结果表明:以正己烷:二氯甲烷和正己烷:乙酸乙酯为提取剂时,各种农药的提取加标回收率偏低;以正己烷:丙酮为提取剂时,提取出的基体杂质较多,对后续净化过程和仪器分析的影响较大;而以乙酸乙酯为提取剂时,提取效率最佳。因此,实验中采用乙酸乙酯作为蜂蜜试样中多种农药残留的提取剂。
2.2 EI/MS 碎片离子的断裂机理与结构
为了准确识别EI/MS的特征离子与试样基体、固定相流失或隔垫流失所产生的干扰离子,特对拟除虫菊酯类农药氯氰菊酯、有机磷类农药乐果和氨基甲酸酯类农药仲丁威的EI/MS碎片离子的断裂机理、结构及其相对丰度进行初步解析。
2.2.1 氯氰菊酯碎片离子的断裂机理与结构

图1 是氯氰菊酯的EI/MS 谱图。由图1可以看出该农药主要的特征离子是m/z为181,163和165的离子。图1 中m/z 为51, 77 ,91 ,115, 127 ,152, 163 ,181, 191, 208 的碎片离子的断裂机理、结构及其相对丰度的初步解析结果见图2及表1。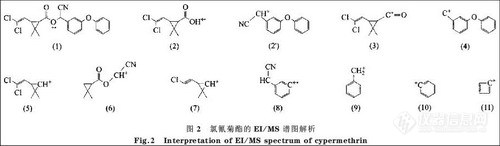
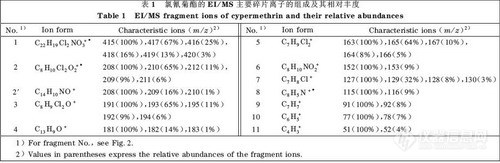
图2中离子(1)是氯氰菊酯的分子离子,由于其在EI源中不稳定,所以相对丰度很弱。离子(2)是离子(1)经过重排脱去中性分子后的奇电子离子,离子(2’)是离子(1)半异裂后的碎片离子;按照图2 和表1的解析结果可以确定是以离子(2’)为主。其他的碎片离子基本上都是离子(1)和离子(2)的碎片离子,还存在断裂机理比较复杂的碎片离子(6)和(8)等。
2.2.2 乐果碎片离子的断裂机理与结构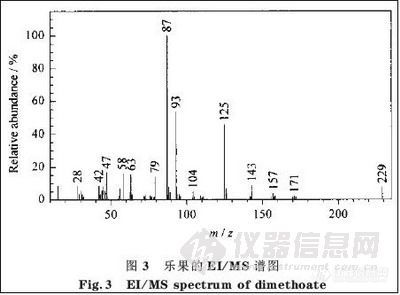
图3是乐果的EI/MS 谱图。由图3 可以看出该农药的主要特征离子是m/z 为125, 93, 和87的离子。图3 中m/z为28, 42, 47, 58, 63 ,79 ,87, 93, 104, 125, 143, 157, 171的碎片离子的断裂机理、结构及其相对丰度的初步解析结果见图4 及表2。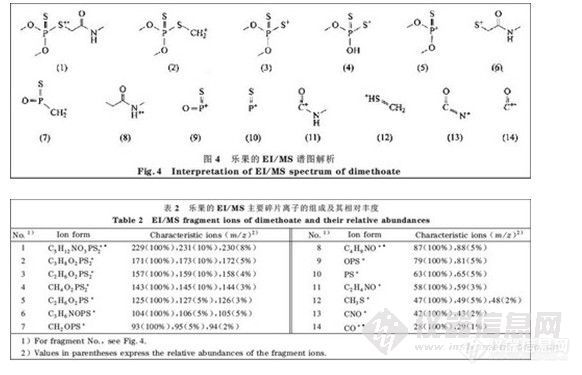
图4中离子(1)是分子离子,离子(2 )、(3)、(5 )、(6)和(11)是离子(1)半异裂后的碎片离子,其他的碎片离子基本上都是离子(1)经过断裂机理比较复杂的重排后的碎片离子复杂的

-
+关注
私聊
-
yhl-87_
第85楼2009/11/06
农药残留检验方法系列讲座(64)(下)气相色谱-质谱法分析蜂蜜中的多种农药残留
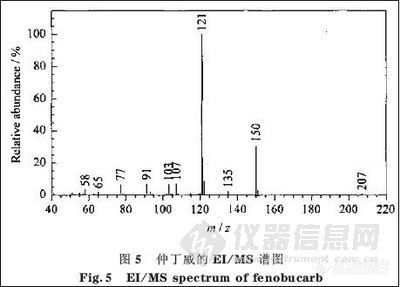
2.2.3. 仲丁威碎片离子的断裂机理与结构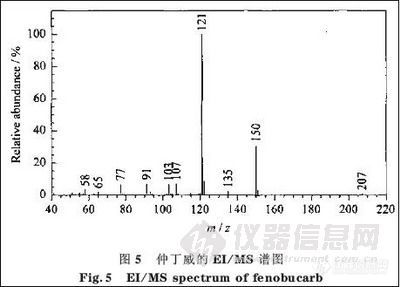
图5是仲丁威的EI/MS 图,由图5可看出该农药的主要特征离子是m/z为150,121的离子。图5 中m/z为58,65,77,91,103,107,121,135,150,207的碎片离子的断裂机理、结构及其相对丰度的初步解析结果见图-6及表3。
图- 6中离子(1)是分子离子,在EI 源中不稳定,所以其相对丰度很弱;离子(2)是离子(2)经过重排脱去中性分子后的奇电子离子;离子(3)、(4)、(5)、(6)都是离子(2)的碎片离子;其他碎片离子基本上都是离子(1 )经过断裂机理较复杂的重排后的碎片离子。
2.3 GC-EI/MS SIM分析
蜂蜜试样的基体较为复杂,提取、净化和浓缩后的提取液采用全扫描方式(SCAN)下的GC-EI/MS分析时,得到的GC-EI/MS SCAN总离子流(TIC)色谱图较为复杂,干扰峰较多;若采用选择离子监测(SIM)方式下的GC-EI/MS 分析,由于GC-EI/MS SIM只对分析物的特征离子进行选择分析,不但分析物的SIM色谱峰强度增大,而且还消除了试样基体中其他共存组分产生的影响,极大地提高了分析方法的选择性和灵敏度。
23种农药的GC-EI/MS分析的保留时间(tR)及特征离子的选择结果见表4
图7为蜂蜜试样添加23种农药混合标准溶液和内标物的GC-EI/MS SIM色谱图。从图7可以看出,经过乙酸乙酯超声提取、Florisil硅藻土净化和前述正己烷-乙酸乙酯混合溶剂洗脱后,加标蜂蜜试样中的23种农药及其异构体和内标物的色谱峰都能达到很好的分离,而且干扰峰非常少。从图7还可以看出:除了抗蚜威农药(峰7)外,其余氨基甲酸酯类农药都存在保留时间和强度都不同的两个色谱峰,如峰(1)和峰(1’)、峰(2)和峰(2’)、峰(3)和峰(3’)、峰(5)和峰(5’)、峰(9 )和峰( 9’);若从GC-EI/MS SCAN TIC色谱图( 图略)中观察,两个TIC色谱峰的EI/MS谱图也不完全相同,其中从保留时间较长的TIC色谱峰(1’),(2’),(3’),(5’)和(9’)的EI/MS谱图中可以观测到分子离子峰及其他碎片离子,而保留时间较短的TIC色谱峰( 1 ),( 2 ),(3),(5)和(9)的EI/MS谱图中均不存在分子离子峰,此现象与贾薇等[9]报道的结果一致。有机磷类农药大都只有一个SIM 色谱峰;拟除虫菊酯类农药中氯菊酯( 峰20)、氯氰菊酯(峰21)、氰戊菊酯( 峰22)分别存在2个、4 个、2 个异构体的SIM色谱峰,以它们各自所有异构体的色谱峰面积之和进行定量分析。
2.4 线性方程、相关系数与方法的检测限
分别取5种不同浓度( 相当于蜂蜜试样中的含量为10.0~500.0μg/kg的23种农药和内标物的混合标准溶液各1.00μL进样,分别采集GC-EI/MS SIM色谱图,以分析物与内标物的峰面积比值(y)对分析物与内标物的含量比值(x)做线性回归分析。
方法的检测限(LOD)在取样量1.00g,定容体积1.00mL、进样体积1.00μL的条件下,以信噪比( S/N)≥3 计算。
线性回归结果表明,在10.0~500.0μg/kg范围内23种农药都呈现良好的线性关系,r 为0.995~0.999,23 种农药的检测限均小于10.0μg/kg。具体数据列于表)。
2.5 加标回收率及相对标准偏差
称取1.00g空白蜂蜜试样( 经分析23 种农药残留均小于LOD)分别添加相当于试样中含50, 100, 200μg/kg的23种农药混合标准溶液,按照“1.2”节所述处理后平行测定5次。分析结果见表5。从表5可看出,对于低加标浓度的试样,有机磷和拟除虫菊酯类农药的平均加标回收率普遍偏高,这是由于试样中的基体组分在分析过程中能够覆盖或掩蔽衬管内表面的部分活性位点,减少了分析物与活性位点之间的相互作用,降低了分析物被吸附或降解的程度,从而使更多的分析物能够进入色谱柱内分离后被检测,增强了检测信号的强度,即产生“ 基体诱导色谱响应增强效应”[10]。随着加标浓度的升高,分析物受该效应的影响逐渐减小,平均加标回收率逐渐趋于正常。23 种农药的3个加标质量浓度下的平均加标回收率均为82%~120%,相对标准偏差(RSD)≤12%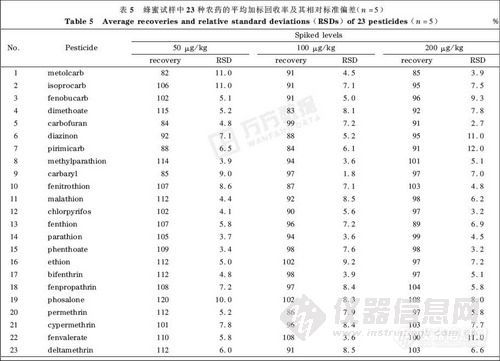
2.6 蜂蜜试样的分析
将本方法应用于蜂蜜试样( 购于当地超市)的分析,结果见表6。在枇杷蜜(Loquat)、枣花蜜(Jujube)、槐花蜜(Scholar)、百花蜜( Flowers)、老蜂农冬蜜(farming)试样中均分析出多种痕量农药残留。在分析出的农药残留中,大多是氨基甲酸酯类农药仲丁威、呋喃丹和抗蚜威等;还分析出有机磷类农药二嗪磷和甲基对硫磷等;对于拟除虫菊酯类农药,甲氰菊酯的检出率较高。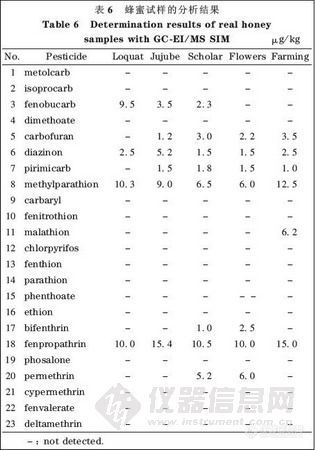
3 结 论
本文将GC-EI/MS SIM方法应用于分析蜂蜜中的23种农药残留,探讨了一些代表性农药的-EI/MS特征离子的结构与断裂机理,采用SIM方法有效地减少了试样基体组分对分析灵敏度和检测限的影响。与其他分析方法相比,具有基体干扰小、选择性高和灵敏度高等优点。分析方法的线性范围、检测限、平均加标回收率和RSD等指标均满足蜂蜜中痕量农药残留的分析要求。
-
+关注
私聊
-
yhl-87_
第86楼2009/11/06
农药残留检验方法系列讲座(65)叶类蔬菜有机氯农药残留测定过程中的提取和净化
摘 要 用柱萃取法提取, 气相色谱2电子捕获检测器( GC-ECD) 测定蔬菜中的多种有机
氯农药. 比较了不同提取溶剂对蔬菜中有机氯农药的提取效率. 就回收率而言, 正己烷和丙酮 (V/ V , 4∶1) > 石油醚和丙酮 ( V/ V , 4∶1) > 二氯甲烷和丙酮 ( V/ V , 4∶1) > 纯正己烷. 用正己烷和丙酮作提取溶剂时, 12 种有机氯农药中有10 种的回收率在80 %以上, 变异系数在1 % ~15 %之间. 不同固相萃取(SPE) 填料对有机氯农药的纯化效率为: 硅胶对目标物质的净化效率要高于硅藻土和氟罗里硅土, 硅胶做填料时变异系数在15 %以下. 因此, 本实验认为用正己烷和丙酮( V/ V , 4∶1) 做提取剂、选择硅胶做填料能够对蔬菜中多种有机氯农药有较好的提取和净化效果, 是一种较理想的蔬菜中有机氯农药的分析方法.。
本文由中国科学院南京土壤研究所的郜红建、蒋 新、王 芳、卞永荣、赵振华、王代长、和焦文涛等研究人员所完成的国家杰出青年科学基金和国家重点基础研究发展规划项目的部分内容,摘自《环境化学》第23 卷 第5 期,现全文介绍如下。
前 言
蔬菜中有机氯农药的测定过程中, 样品的提取和纯化过程是前处理成败的关键. 蔬菜中有机氯农药的残留可以用正己烷、丙酮、二氯甲烷、石油醚、乙腈、甲醇或以它们不同比例的混合溶剂进行提取[1 —6 ] . 纯化过程可以选择氟罗里硅土、硅胶、硅藻土、活性炭等材料作为吸附介质进行净化[1 , 4 , 7 , 8 ] . 但不同提取溶剂和净化材料的结构和性质各不相同, 其提取和净化效率有很大差别。
本文研究柱萃取法提取蔬菜中的有机氯农药, 并比较不同提取剂对这类农药的提取效率, 不同固相萃取(SPE) 填料的净化效果, 从而选择最佳的提取溶剂和净化材料,为蔬菜中有机氯农药的常规分析提供理论依据
实 验
1 实验方法
1.1 农药和蔬菜
α-HCH , β-HCH , γ-HCH , δ-HCH , p,p’-DDT , p,p’-DDE , p,p’-DDD , o,p’-DDT ,o,p-DDE , 狄氏剂、异狄氏剂和六氯苯标样 (购自德国Dr. Ehrenstorfer 公司) 用正己烷溶解, 配制成1mg·ml - 1的母液, 用时稀释。
青菜矮脚黄( Brassica Chinensis L) 样品采自南京市六合蔬菜基地. 取2.5kg 样品带回实验室后, 用四分法保留1kg , 洗净表面灰尘, 凉干表面水分后粉碎. 取一定量的蔬菜样品放入40ml 离心管, 加入已经配好的有机氯农药的标准混合溶液, 使各种农药在蔬菜中的浓度为100ng·g- 1 . 在涡旋振荡仪上充分混合后, 在室温下密封保存24h.
1.2 样品的提取和纯化
取2.0g 蔬菜样品, 1.0g 石英砂和20.0g 无水硫酸钠, 放入研钵中, 充分研磨后装入玻璃柱(30cm ×1.5cm) . 分别用30ml 正己烷和丙酮.( V/ V , 4∶1) 、石油醚和丙酮.( V/ V , 4∶1) 、二氯甲烷和丙酮.( V/ V , 4∶1) 缓缓淋洗玻璃柱, 将流出液收集在100ml分液漏斗中, 加入10ml 浓硫酸去除提取液中的色素, 10.0ml 2 %的硫酸钠溶液去除水溶性杂质, 将有机相转移到50ml 梨形蒸馏瓶中, 在旋转蒸发仪上浓缩至约1.0ml , 然后通过SPE 柱进行净化.
在SPE 柱中依次加入1.0g 无水硫酸钠, 1.0g 填料 (硅胶、硅藻土和氟罗里硅土)和1.0g 无水硫酸钠. 用8.0ml 石油醚淋洗SPE 柱, 然后将1.0ml 的浓缩提取液转入SPE柱, 最后用10.0ml 石油醚和二氯甲烷 (V/ V , 9∶1) 洗脱. 洗脱液在旋转蒸发仪上浓缩至约1.0ml , 转移到1.0ml 容量瓶中, 用氮气流吹蒸定容, 移入进样瓶中备测.。
1.3 GC-ECD 分析
Agilent 6890/ ECD 63Ni 气相色谱仪, HP-5 色谱柱 (30m ×0.32mm ×0.25μm) . 进样口温度220 ℃; 检测器温度280 ℃; 程序升温: 初始温度60 ℃, 保持1min ; 以20 ℃/min-升到140 ℃, 保持5min ; 然后以12 ℃/min升到280 ℃, 保持4min ; 不分流进样; 载气为高纯氮气, 流速1.5ml·min - 1 ; 柱前压50kPa , 进样量1μl , 外标法定量.
2 不同提取溶剂的提取效率
图1 给出了不同提取溶剂对蔬菜中有机氯农药的提取效率. 从图中可以看出, 正己烷的提取效率较低, 其回收率在8.9 % —83.7 %之间, 大多小于60 % , 只有少数在70 %以上. 这可能是因为有机氯农药与蔬菜组织充分作用后结合较牢, 仅仅用正己烷做提取剂, 由于其疏水性较强, 不能到达蔬菜组织的内部, 从而使它的提取效率较低. 正己烷/ 丙酮.( V/ V , 4∶1) 做提取剂时, 大大提高了提取的效率, 对12 种有机氯农药的提取的回收率在61.4 % —125.8 %之间, 其中, 10 种农药的回收率在80 %以上. 对农药的提取效率依次为: DDT 类> 六氯苯> 六六六类> 狄氏剂和异狄氏剂. 狄氏剂和异狄氏剂的回收率相对较低, 均在70 %以下, 这可能与在提取时用浓硫酸磺化法去除提取液中的色素, 使部分狄氏剂和异狄氏剂损失所致. 石油醚和丙酮.(V/ V , 4∶1) 做提取液,其提取效率优于(或高于) 正己烷, 而低于正己烷/ 丙酮. 绝大部分物质的回收率在70 %以下, 只有少数物质的回收率高于80 %. 而二氯甲烷和丙酮.(V/ V , 4∶1) 的提取效率比石油醚和丙酮.( V/ V , 4∶1) 还要低. 综上所述, 不同提取溶剂对蔬菜中有机氯农药的提取效率为: 正己烷和丙酮.(V/ V , 4∶1) > 石油醚和丙酮.( V/ V , 4∶1) > 二氯甲烷和丙酮.( V/ V , 4∶1) > 正己烷. 因此, 本实验认为正己烷/ 丙酮(V/ V , 4∶1) 对蔬菜中的多种有机氯农药有较好的提取效果.
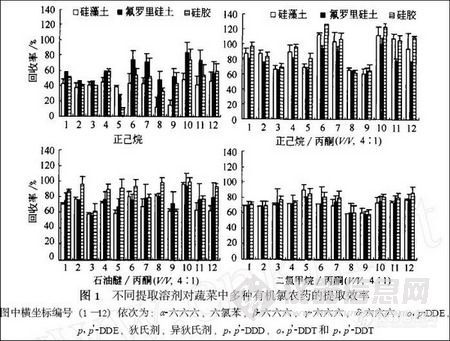
3 不同萃取材料的净化效率
固相材料性质不同的固相萃取柱, 对环境样品的富集和净化效率也有不同. 不同固相萃取(SPE) 填料对12 种有机氯农药的纯化效率为: 硅胶> 氟罗里硅土> 硅藻土(图1) . 不管选择何种提取剂, 硅胶对目标物质的净化效率要高于硅藻土和氟罗里硅土, 且硅胶做填料时其相对标准偏差较低, 在15 %以下. 因此, 选择硅胶做填料能够对多种有机氯农药达到较好的净化效果.
4 分析方法的可靠性
为了验证本实验所得结果的可靠性, 选择正己烷/ 丙酮 (V/ V , 4∶1) 作提取溶液,用硅胶做固相萃取柱的净化材料, 测定12 种有机氯农药添加浓度为200ng·g- 1的蔬菜样品, 其提取和净化效率与国家标准方法[9 ]和美国EPA 的方法[10 ]进行比较, 其结果见表1.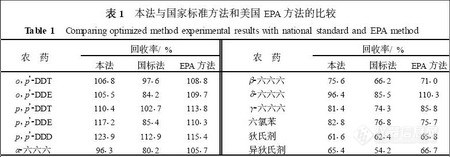
从表中可以看出, 本实验有机氯农药的提取和净化方法要优于国标法, 表现为回收率普遍高于国标法. 与美国EPA 的方法相比, 其回收率基本上与之接近, 其中β-六六六和六氯苯的回收率还略高于EPA , 说明本实验所得到的方法对蔬菜中痕量有机氯农药的定量分析是准确可靠的。
-
+关注
私聊
-
yhl-87_
第87楼2009/11/06
农药残留检验方法系列讲座(66)气相色谱一质谱、电子捕获、氮磷检测器联用技术分析商品农药有效成分
摘 要 建立了气相色谱一质谱、电子捕获、氮磷检测器联用技术分析商品农药有效成分的方法。样品由乙腈、正己烷溶解稀释定容后,由气相色谱一质谱、电子捕获、氮磷检测器联用技术定性分析和确证。该方法具有简单、快速、可靠的特点,适用的商品农药有效成分范围包括有机磷、有机氯、菊酯、氨基甲酸酯和有机氮类等非受热易分解农药。
本文由东莞市农业检验监测所的李亮、昊春梅、陈健航、张少丰等人共同完成,第一作者是从事农产品质量安全监督检测工作 他的联系方式Email: mycolity@163 现全文介绍如下。
前 言
农药的有效成分是农药质量的关键控制指标,决定农药的效用和用途。当前的农资市场存在部分商品农药无标识有效成分,标称有效成分与实际有效成分不一致,在名为高效低毒的农药中混配高毒高残留成分,本应单一成分的农药加入多种成分提高杀虫效果等不规范及假冒伪劣的情况。引发治虫无力,药害,农民在不知情的情况下把高毒农药喷施到蔬菜水果上,造成残留农药超标和食用中毒的后果。严重损害农民利益,威胁农业生产安全和农产品质量安全。
目前已有不少商品农药有效成分的检测方法,但多数只是从该商品农药的有效成分含量是否达到标准要求值的角度去分析.,遇到诸如高效低毒的菊酯类农药中混配有高毒有机磷农药,标称为有机磷农药的商品中混有菊酯类或氨基甲酸酯类农药等情况时,这些方法的局限性就凸显了出来。所以开发可实现多有效成分或完全未知有效成分商品农药的分析方法非常迫切。本文以气相色谱仪 (GC) 和质谱检测器 (MS)、微电子捕获检测器 (μ-ECD)、氮磷检测器 (NPD) 联用,实现了大范围类别商品农药有效成分的快速定性分析。
实 验
1 实验部分
1.1主要仪器
GC-6890N一5975B气相色谱-质谱仪 (带自动进样器,配三路分流器Three—Way Splitter Kit,G3183B,微电子捕获检测器“μ-ECD,氮磷检测器NPD):美国安捷伦-(Agilent)公司产品,配MSD ChemStation工作站,NIST05谱库;HS501 D往复式振荡机 (IKA—WERKE);Sigma 3-16离心机 (美国Sigma);Dispensette Organic 5~50mL瓶顶分液器 (德国BRAND);l、5、10mL移液枪 (德国Eppendorf);50mL带螺纹盖聚丙烯离心管;分析天平(精度0.00019)。
1.2主要材料与试剂
乙腈:HPLC级(德国Merck);正己烷:HPLC级(德国Merck);氯化钠:分析纯,140℃烘烤4h。
农药标准品:由中国标准技术开发公司标样开发部提供,浓度均为100μg/mL的有证标准溶液。根据需要配制成所需混合标准溶液。
1.3标准溶液的配制
使用前吸取适量原液 (浓度为l00μg/mL)用正己烷稀释成所需浓度的单种组分标液或混合多组分标液即可。
1.4样品处理。试样溶解或稀释
准确称取商品农药0.3000g至50mL聚丙烯离心管中,用移液枪加人2.00mL乙腈,轻轻摇动离心管让农药与乙腈混溶,然后用瓶顶移液器加入47.7mL正己烷,盖好后置于往复式振荡机剧烈振摇10min。加入1g氯化钠,再振摇1min,放人离心机中,以3000r/min离心5min。吸取上层液0.50mL放置于另一支50mL离心管中,加人49.5mL正己烷,盖好放于振荡机上剧烈振荡5min。移取适量上机测定。
1.5仪器条件
1)色谱条件:色谱柱:DB一1701石英毛细管柱(30m×0.25mm×0.25μm);升温程序:初始温度80℃,以30℃/min的速率升至280℃,保持15min;进样口温度290℃;载气(高纯氦气)流速:1.2mL/min;不分流进样,进样量2μL。
2)质谱条件:电子轰击(EI)离子源;电子能量70eV;离子源温度230℃;接El温度280℃;溶剂延迟时间3.5min;辅助EPC(Aux EPC)的压力为3.8psi。
3)微电子捕获检测器条件:温度300℃;尾吹:高纯氮气60mL/min。
4)氮磷检测器条件:温度330。C;高纯氢气3mL/min;空气60mL/min;尾吹:10mL/min。氢气开启时间:3.5min。
2结果与讨论
2.1分析效果
在116个商品农药样品中,除了阿维菌素、苏云金杆菌等要分别使用液相色谱法和其他检测途径才能进行分析外,大部分的商品农药的标称有效成分 (有机磷类、有机氯类、菊酯类和氨基甲酸酯类或有机氮类农药中的非受热易分解部分) 都能使用上述方法进行定性分析及确证。有些商品农药未标有效成分,用此法能分析鉴定出其有效成分;有些商品农药标注的有效成分与所含的有效成分不一致也能发现。我们曾在一些标注高效低毒的农药中检测出含较高浓度的甲胺磷、治螟磷、甲基异柳磷等高毒,高残留有效成分;菊酯类农药制剂中加入有机磷类或氨基甲酸酯类农药等,为农资打假、农业生产及农产品质量安全管理提供了快速且极具参考价值的数据。

-
+关注
私聊
-
yhl-87_
第88楼2009/11/06
农药残留检验方法系列讲座(66)(下)气相色谱一质谱、电子捕获、氮磷检测器联用技术分析商品农药有效成分
2.2实现GC与MSD、μ-ECD、NPD联用的技术及装置
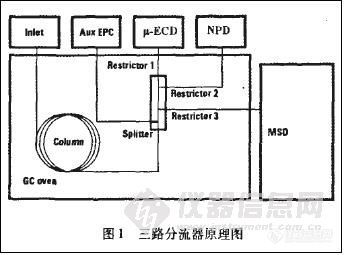
实现GC与MSD、μ-ECD、NPD联用的装置为Agilent三路分流器Three—way Splitter Kit,G3 1 83 B。
其技术原理见图1。该分流器 (Splitter) 被安装在毛细管色谱柱的出口端与检测器之间,从色谱柱流出来的分析物通过它分成三路,分别接入MSD、μ-ECD、NPD中,通过安装不同内径和长度的限流管 (Restrictor) 结合辅助电子压力控制器 (Aux EPC) 的压力调节,能够实现调节进入不同检测器的流量。本文调节相关部件的参数使进入三个检测器的流量比为1:1:1,流出化合物在各检测器的保留时间一致。(该工作由安装工程师完成,更具体的原理和安装调节方法参见产品说明书。)
2.3有效成分的定性和确证
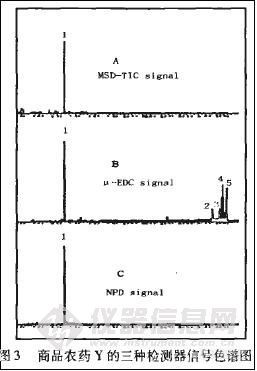
对商品农药的有效成分进行分析前,虽然有时可根据其标签或登记信息知道其大致的有效成分及含一量,但是在现实的分析中,样品的有效成分应看作未知,以便得到更客观全面的分析结果。就这一点来说,使用质谱检测器分析很有优势,因质谱检测器为通用型检测器,其全扫描 (SCAN) 对绝大部分的有机物有响应,还能提供该物的结构信息。但商品农药中的杂质品种多且含量往往很高,这造成了大部分样品的总离子流 (TIC) 图出峰多且复杂,无法分清那些峰是有效成分的峰,只能逐一地对色谱峰进行扣背景和检索,这种毫无主次的工作极其耗费时间且常常会漏掉本属于主要有效成分的色谱峰,如图2中的图AMSD—TIC signal为一种商品农药的总离子流图。
另外,质谱检测器很容易受到高浓度样品的污染,所以在用其进行检测时,样品应该尽可能被稀释到主成分(包括主要杂质)的浓度很小。但样品一旦被稀释到浓度很小,质谱的SCAN功能的灵敏度往往无法满足要求。
用ECD、NPD、MSD与GC联用技术就可以解决以上问题,因为电子捕获检测器(ECD)是灵敏度最高的气相色谱检测器,它仅对那些能捕获电子的化合物,如卤代烃,含N、P和S等杂原子的化合物有响应,它灵敏度高,选择性好。氮磷检测器NPD对氮、磷化合物具有专一性,且在所有的检测器中对N、P化合物的灵敏度最高[2]。这两种检测器分别对商品农药中的有机氯类、菊酯类有选择性的响应;对有机磷类、氨基甲酸酯类 (或有机氮类) 有效成分有选择性的响应;对其它杂质响应极小或没有响应。所以大部分商品农药的色谱图都比较干净,寻找谱图中商品农药的有效成分时能够一目了然地发现有效成分的色谱峰。该峰的保留时间与TIC图中该化合物的保留时间一致,根据ECD和NPD的谱图快速地在TIC图中找到对应的色谱峰进行扣背景,从而迅速得到对应化合物的质谱图,作谱库检索或解释,实现定性确证。有时因为灵敏度不够TIC图中没有峰,可根据ECD和NPD谱图提供的线索大致确定该化合物的范围,然后使用SIM功能在该保留时问段设定被怀疑的目标物的特征离子重新进样分析采集谱图。最后利用SIM图对有效成分进行确证。
例如图2为商品农药x的三种检测器信号图,该农药的质谱检测器总离子流图(A MSD—TIC signal)有大量的色谱峰,甚至很多峰是连片出现,如果逐个地进行扣背景检索,一个样品就要耗费半天的时间,且无法区分哪个地方是有效成分的峰,哪个地方是杂质峰或背景干扰峰。由于样品中的有效成分未知,不能确定目标化合物,无法用选择性扫描(SIM)。但通过ECD信号 (图2中Bμ—ECD signal) 和NPD信号 (图2中C NPD signal) 可以明显地看出:该商品农药共有三种有效成分。由ECD和NPD信号图的色谱峰初步确定有效成分的保留时间,在对应TIC图中该时间处进行有针对性的扣背景及谱库检索后发现,标号为l的色谱峰是敌敌畏农药,标号为2和3的色谱峰分别是氯氰菊酯的两个同分异构体。正反检索匹配率均达90%以上,该商品农药的有效成分便得到了初步确证。为了得到更确切的结果,按1.3方法配制敌敌畏和氯氰菊哺混合标液上机,得到敌敌畏、氯氰菊酯的标准色谱图、质谱图,拿其保留时间、质谱图与样品中的1、2、3号峰的保留时间、质谱图进行对比,均符合确证所需的匹配要求,结果得到确证。
又如图3为商品农药Y的三种检测器信号色谱图,如果只用质谱检测器进行全扫描,该农药的总离子流图(图3 A MSD—TIC signal)中显示有一个峰 (图中的1号峰),经谱库检索,知其为乐果,这与该农药所标注的有效成分一致。但再观察分析μ-ECD的信号图,发现还有2、3、4、5号峰,根据经验推测这可能是一种菊酯的多个同分异构体,在A信号图对应的地方进行扣背景后检索,未找到匹配率高的物质。据经验推测该物质可能是氯氰菊酯,尝试在该保留时间处设定氯氰菊酯的特征离子,分别进氯氰菊酯标准溶液和重新进样分析后,样品的特征离子及丰度比例与标液的一致。则该商品农药含氯氰菊酯得以确证。
3 结论
又如图3为商品农药Y的三种检测器信号色谱图,如果只用质谱检测器进行全扫描,该农药的总离子流图 (图3 A MSD—TIC signal) 中显示有一个峰(图中的1号峰),经谱库检索,知其为乐果,这与该农药所标注的有效成分一致。但再观察分析μ-ECD的信号图,发现还有2、3、4、5号峰,根据经验推测这可能是一种菊酯的多个同分异构体,在A信号图对应的地方进行扣背景后检索,未找到匹配率高的物质。据经验推测该物质可能是氯氰菊酯,尝试在该保留时间处设定氯氰菊酯的特征离子,分别进氯氰菊酯标准溶液和重新进样分析后,样品的特征离子及丰度比例与标液的一致。则该商品农药含氯氰菊酯得以确证。
本文建立了以GC—MS、μ-ECD、NPD联用技术,快速分析商品农药有效成分的方法。该方法前处理过程及仪器分析均简单易操作,得到结果迅速,灵敏度高,定性确证结果可靠,适用商品农药范围广,较全面。可用于商品农药真伪辨别和功能成分分析解剖,农资打假,农业药害类事故的调查分析等方面。
-
+关注
私聊
-
yhl-87_
第89楼2009/11/06
农药残留检验方法系列讲座(67)中草药中有机磷及氨基甲酸酯类
农药残留量的GC - MS 测定
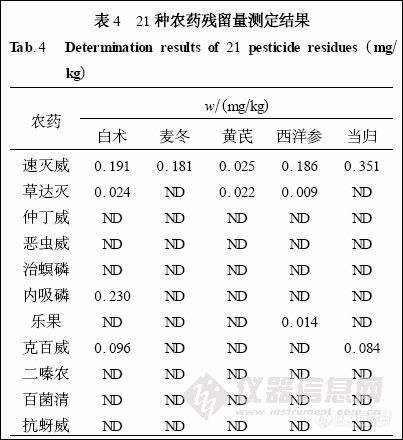
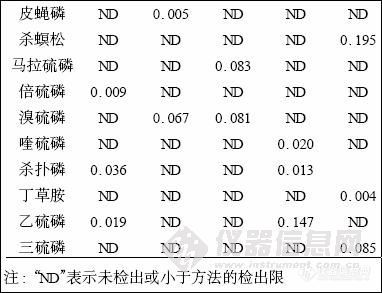
摘 要: 气相色谱-质谱法同时测定中草药中多种有机磷及氨基甲酸酯类农药残留量。采用V (乙腈) ∶V (丙酮) = 3∶7 混合溶剂微波辅助提取, 弗罗里硅土和中性氧化铝层析柱净化, 气相色谱-质谱( GC-MS) 联用检测, 农药混标在0.01~1.0μg/mL 范围内线性良好, 在0.5、0.1、0.05μg/mL 3 个水平添加平均回收率分别为86.5 %~110.6 %、81.2 %~108.3 %和72.9 %~122.3 % , 相对标准偏差分别为2.6 %~8.3 %、4.6 %~9.7 %和2.3 %~10.7 %。
本文由万益群1 ,2 , 李申杰2 , 付贵琴2 (1. 南昌大学食品科学教育部重点实验室,;2. 南昌大学分析测试中心,) 共同完成的江西省自然科学基金和南昌大学食品科学教育部重点实验室开放基金资助项目,属于长江学者和创新团队发展计划中的部分内容。现全文介绍如下
前 言
中草药由于需求量大, 约有80 %是靠人工种植的。为抵抗病虫害侵袭, 人们在种植过程中往往要喷洒农药, 这就使中药材受到农药的污染。农药残留问题也成了制约中草药出口的一大障碍,因此, 加强中草药中农药残留量的检测方法研究十分必要。近年来, 对中草药中农药残留量检测常有报道, 但多是对某一类农药残留量的检测,对多类农药残留量同时检测的不多, 而同时检测不同种类的多种农药残留量是今后研究的一个趋势。本文较传统分析方法, 提高了分析速度, 简化了操作步骤, 节约了试剂。已应用于实际样品分析。
实 验
1 实验部分
1. 1 仪器
Agilent 6890N 型气相色谱仪 (美国) , 配备Agi2lent 5973i 质谱仪和Agilent 7683 自动进样器; NIST图谱检索库 (13 万标准质谱图) ; 粉碎机 (上海嘉定仪器有限公司) ; RE - 52A 旋转蒸发仪 (上海亚荣生化仪器厂) ; Milestone ETHOS E 微波溶样器 (意大利) 。
1. 2 标准品和试剂
农药标准品: 速灭威, 草达灭, 仲丁威, 恶虫威, 治螟磷, 内吸磷, 乐果, 克百威, 二嗪农, 百菌清, 抗蚜威, 皮蝇磷, 杀螟松, 马拉硫磷, 倍硫磷, 溴硫磷, 喹硫磷, 杀扑磷, 丁草胺, 乙硫磷, 三硫磷。以上21 种标准品由农业部环境保护科研监测所研制(浓度均为100μg/mL) 。白术等中草药(市售) ; 正己烷、二氯甲烷、丙酮、乙腈等试剂均为分析纯, 使用前均重蒸; 无水硫酸钠在650 ℃马弗炉中灼烧4 h , 备用; 弗罗里硅土和中性氧化铝在650 ℃马弗炉中灼烧3 h , 然后在130 ℃活化后使用。
1. 3 实验方法
1. 3. 1 样品的提取
药材用粉碎机粉碎后过0. 45mm 粒径筛。准确称取1. 0 g 样品于萃取罐中, 加入15 mL V (乙腈) ∶V (丙酮) = 3∶7 溶液, 混匀, 在100 ℃下微波萃取10 min , 萃取液转移至250 mL烧瓶中。然后用10 mL 提取液分两次洗涤萃取罐,合并入250 mL 烧瓶中, 在旋转蒸发仪上浓缩至约1 mL 。
1. 3. 2 样品的净化
层析柱自下而上装填: 1 cm无水硫酸钠, 5 g 弗罗里硅土, 3 g 中性氧化铝, 1cm无水硫酸钠。用20 mL 正己烷预淋洗柱子, 然后将浓缩液转移至柱头, 用10 mL V (二氯甲烷) ∶V (丙酮) = 9∶1 分多次洗涤浓缩瓶倒入柱中, 再用10 mL V (二氯甲烷) ∶V (丙酮) = 1∶1 和10 mL V (二氯甲烷) ∶V (丙酮) = 1∶9 洗脱。收集洗脱液于250mL 烧瓶中, 在40 ℃水浴下旋转蒸发至干, 用正己烷定容至1 mL , 待测。
1. 3. 3 色谱、质谱条件
GC: HP25MS 毛细管柱 (30 m ×0. 25 mm i . d. , 0. 25μm) ; 色谱柱温度: 初温50 ℃, 保持1 min , 以25 ℃/min 升至150 ℃, 保持2 min , 再以2 ℃/min 升至180 ℃, 再以15
℃/min升至190 ℃, 最后以20 ℃/min 升至230 ℃,保持5 min ; 进样口温度: 250 ℃; 载气: 高纯氦气 (1 mL/min , 恒流) 。MS: 溶剂延迟5 min ; 扫描范围: 50~500 u ; 离子源温度: 230 ℃; 四级杆温度:150 ℃; 检测器接口温度: 280 ℃; EI 电子能量:70 eV; 进样体积1μL ; 外标法定量。
2 结果与讨论
2. 1 离子通道的建立
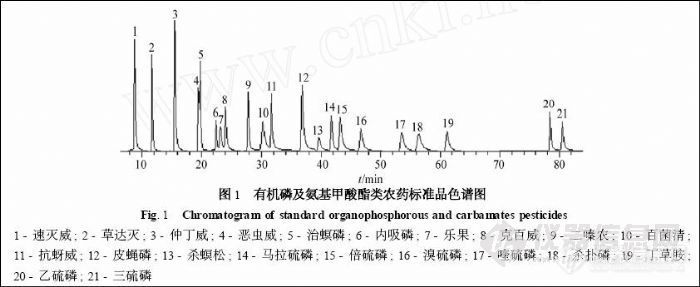
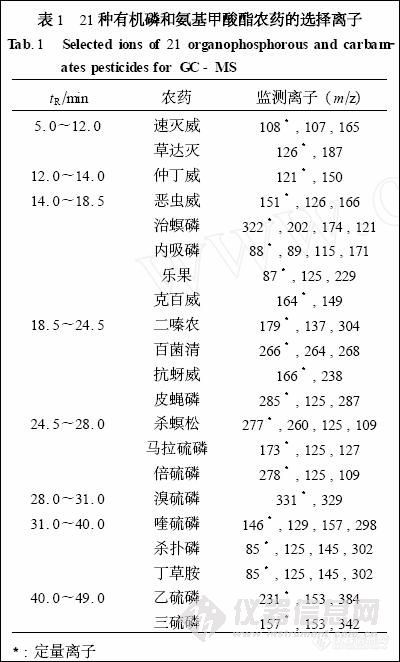
根据21 种农药的TIC 图 (见图1) , 将保留时间相近的农药分为一组, 为一个通道。本实验设计了8 个离子通道, 各离子通道中选择监测离子见表1。
2. 2 微波提取条件的选择
采用微波提取, 选用提取温度、提取时间、溶剂用量和溶剂种类4 个因素, 每个因素取3 个水平, 按L9 (34 ) 正交设计表 (见表2) 进行实验。最后确定微波萃取的条件为: 在100 ℃下加入15 mL V (乙腈) ∶V (丙酮) = 3∶7 混合溶剂萃取10 min。
2. 3 净化条件的选择
采用Florisil 和中性氧化铝混合装柱。洗脱剂用V (正己烷) ∶V (二氯甲烷) = 7∶3、V (二氯甲烷)∶V (丙酮) = 7∶3 和V (丙酮) ∶V (甲醇) = 1∶1 进行了对比实验, 结果发现正己烷:二氯甲烷洗脱不完全, 二氯甲烷:丙酮则需要55 mL 才能洗脱完全,丙酮:甲醇洗脱下来的杂质太多。通过实验最后确定分别用10 mL V (二氯甲烷) ∶V (丙酮) = 9∶1、1∶1
和1∶9 的混合溶剂洗脱, 淋洗完全且杂质较少。
2. 4 标准曲线及线性范围
将混合农药标准贮备液(1.0 μg/mL) 配制成0.01、0.05、0.1、0.2、0.5、1.0μg/mL 的标准工作液, 按选定的色谱、质谱条件进行实验, 21 种农药在0.01~1.0μg/mL 的范围内线性关系良好, 相关系数在0.996~0.9998 之间, 检出限在0.001~0.024μg/mL 之间。
2. 5 回收率及精密度的测定
取白术样品1. 0 g , 分别加入500、100、50μL的混合农药标准贮备液(1. 0 ng/μL) , 放置一段时间, 让农药充分渗透至样品中, 按选定的实验方法进行提取、净化和检测, 每个水平重复实验5次, 根据定量选择离子峰面积计算各种农药在3个添加水平的平均回收率和精密度, 结果见表3。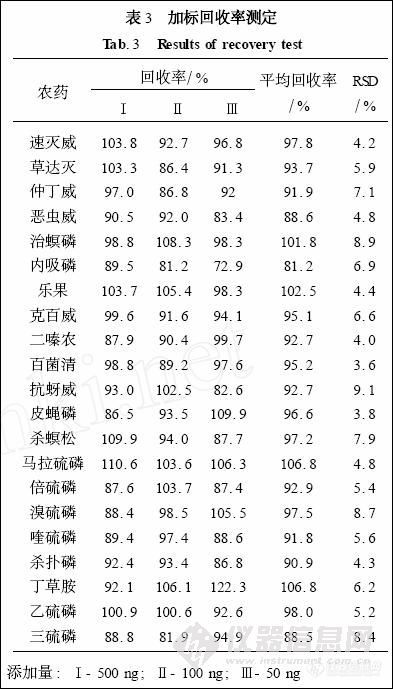
2. 6 样品检测
称取一定量已制备好的白术、麦冬、黄芪、西洋参和当归样品, 按选定的实验方法进行提取、净化和气相色谱2质谱测定, 结果见表4。从分析结果看出, 速灭威在5 种中草药中均有检出, 其他农药也有的能检出, 但其量均较低。