-
+关注
私聊
-

geebzbz
第20楼2010/11/23
前面的那段峰,我是不需要的,也就是在11min左右之前的我都可以不用考虑。
之前我做过0%-100%甲醇的梯度洗脱的,图如下: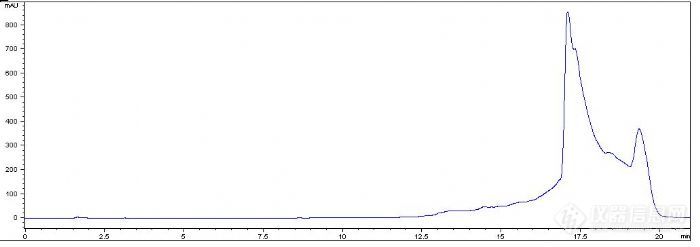
另外,在做完样品之后,也做了一个空白,图如下(当时没再重复,也不知道4-6min的那些波动是不是残余的样品):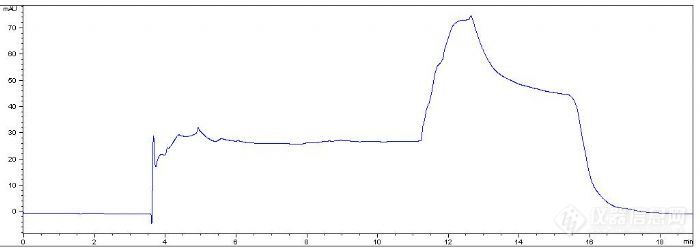
结合这些图,各位再帮忙分析分析下,谢谢大家!有水有渝(xky0230699) 发表:楼主做一个空白看看是样品引起的基线波动还是梯度引起的,做这个不使用对照品,只是归一法看一下出峰吗?8-13分钟这一段洗脱的都不要了吗,如果要的话这个直接跳跃式的梯度肯定是不行的。建议把色谱柱用梯度多清洗几遍,拿一其它物质先测一下柱效,看色谱柱是否正常,还有你的梯度方法使用的流动相你优化过了吗,做过0%-100%的梯度结果谱图是怎么样的,色谱峰形好吗?感觉上流动相的组成可能也不合理。