-
+关注
私聊
-

guizi1985
第19楼2011/06/21
用乙腈提取,涡旋2min,超声15min,振荡15min,乙酸锌沉淀蛋白,氮气吹干后10%甲醇3ml溶解残渣,过HLB柱
活化:3ml甲醇,3ml水平衡, 上样,10%甲醇水3ml淋洗,甲醇3ml洗脱,吹干1ml定容,结果: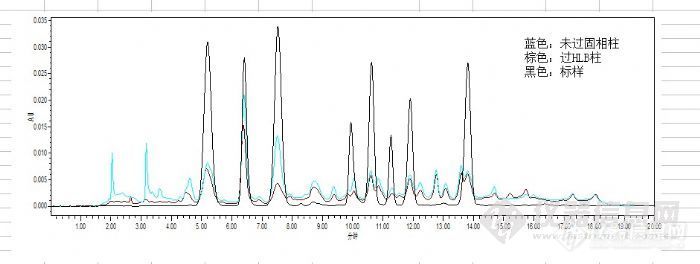
过柱方法采用了HLB通用过柱方法,杂质没洗掉,3号主要药物洗掉一大半,是不是淋洗应该好好摸索啊?
我估计上样时就有药物掉下来了!溶解残渣是不是应该酸化上样?水中月(lianlxh) 发表:希望看到你过完HLB柱子的色谱图。前段时间也做药物残留,基质比较复杂,所以大多数的都需要过SPE柱子的,即使这样也是有很多无法能够完全分离的。还有就是如果你做混标的回收率的话比较不好达到要求,如果一个个添加的话就比较好做,这个具体什么原因我也说不明白。