-
+关注
私聊
-

chaifa
第17楼2011/08/15
亲,从你的图上看,我觉得主峰前面的那个小峰有可能是基线噪音造成的,你看看前面的峰都快赶上主峰了,呵呵,Y轴量程才12点多主峰又一点点,说明你进样量少的多,很有可能是因为你在自动进样每一针之间没有设置洗针所致,当然因为是20分种时间比较长,没有柱温对控温上也是很难做到,如果柱温不断的变化也有这种情况的出现。解决方法,一、加上一个高于你环境温度的柱温看看。二、在每进一针后设置上洗针,三、走一个流动相的空白看看,如果主峰前也有一个小峰,可以走样时加入梯度洗脱进行分离,四、把柱子反过来用下(原则上不允许),五、管路或检测器中有气泡而出现鬼峰,六、就是最让人骂的办法————更换柱子,好了,就这么多了,愿你解决后给我们回个话,告知。
305039484(305039484) 发表:各位大侠:
我现在使用C18柱反向色谱分离目的物质,流动相是乙腈:水(0.25ml磷酸加入重蒸水调节PH约为2-3)=20.5:79.5,系统平衡了近一个小时后进第一针样,峰形如下:目的峰分离时间为20.119min,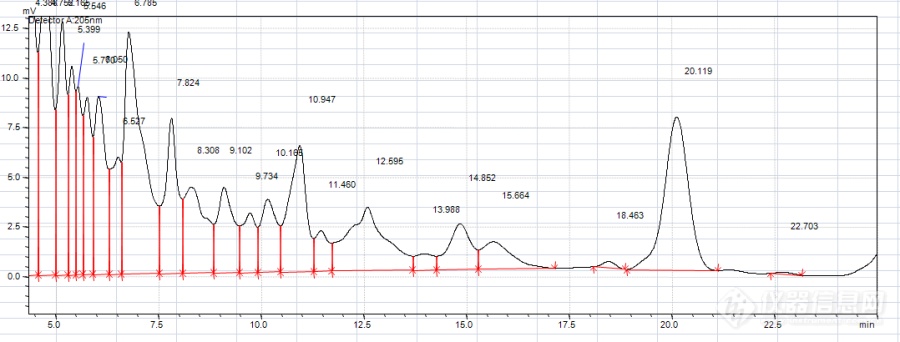
进的针数多了,峰形慢慢的变形,如下图: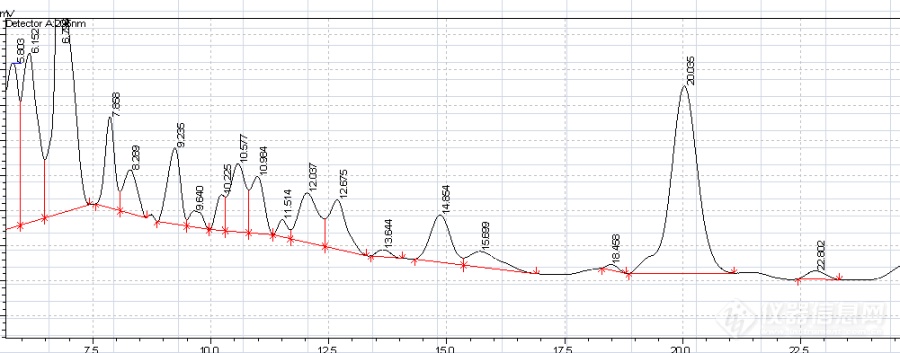
最后在主峰前面逐渐分化出一个小峰。如下图: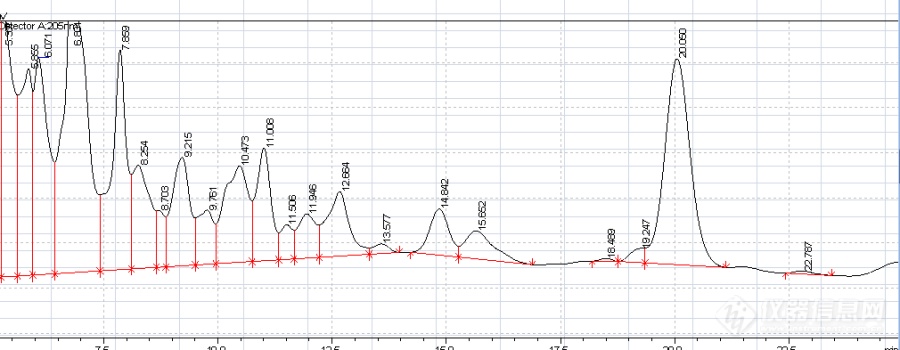
我想请教各位大侠,是什么原因使峰这样变形了,是不是流动相PH不合适?还是柱子本身没平衡好,使本该分离出的小峰没被及时检测出?
-
+关注
私聊
-

305039484
第18楼2011/08/15
我贴的图是放大了的,溶剂峰和2-3分钟的杂质峰量程有100多,我是手动进样的,我的柱温是39度,我的图谱样是生产过程的中间样品,样品比较脏,成分多,基线确实很难是在零点。
chaifa(chaifa) 发表:亲,从你的图上看,我觉得主峰前面的那个小峰有可能是基线噪音造成的,你看看前面的峰都快赶上主峰了,呵呵,Y轴量程才12点多主峰又一点点,说明你进样量少的多,很有可能是因为你在自动进样每一针之间没有设置洗针所致,当然因为是20分种时间比较长,没有柱温对控温上也是很难做到,如果柱温不断的变化也有这种情况的出现。解决方法,一、加上一个高于你环境温度的柱温看看。二、在每进一针后设置上洗针,三、走一个流动相的空白看看,如果主峰前也有一个小峰,可以走样时加入梯度洗脱进行分离,四、把柱子反过来用下(原则上不允许),五、管路或检测器中有气泡而出现鬼峰,六、就是最让人骂的办法————更换柱子,好了,就这么多了,愿你解决后给我们回个话,告知。