-
+关注
私聊
-

雨红星宇
第12楼2018/12/19
我用的pe 的原吸,80ng /ml 的铅标准溶液吸光度才0.11,你这浓度点40,吸光度都有0.6,这应该是不同仪器的差异吧,所以我没法评判你的灵敏度,但是我们的升温程序跟你不一样,我的灰化温度650,原子化温度1500,不同仪器估计升温程序也不能拿来比较,所以楼主自己摸索吧
阳光正好521(m3160671)发表:我的消解是湿法消解,用1+9的硝酸高氯酸混合酸,消解至大概剩余1mL冒白烟时停止,加10ML水,赶酸,赶酸至还剩余大概1ml溶液时停止,用水洗出来定容至10刻度
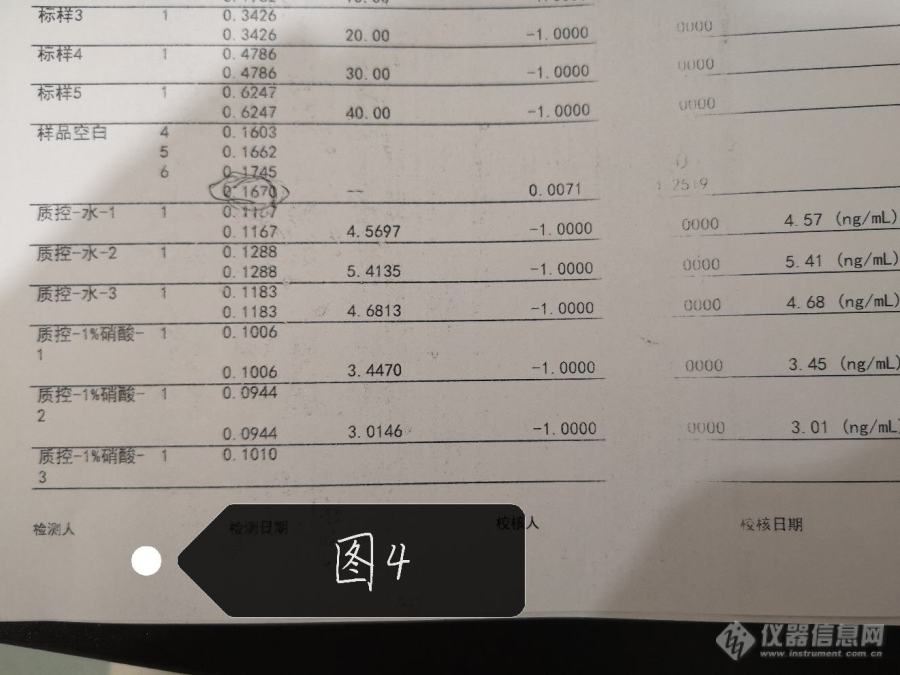
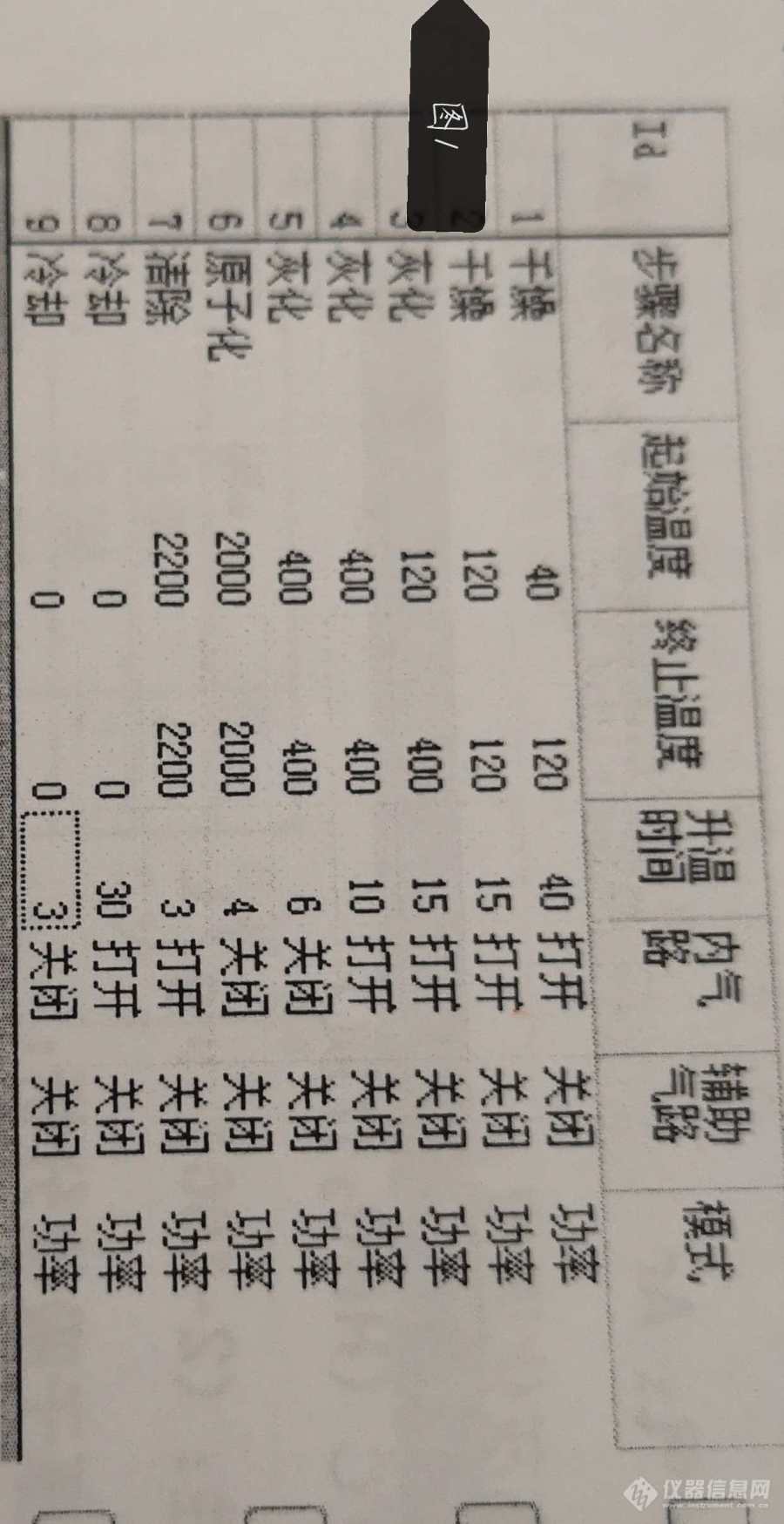
仪器的升温程序为下图1
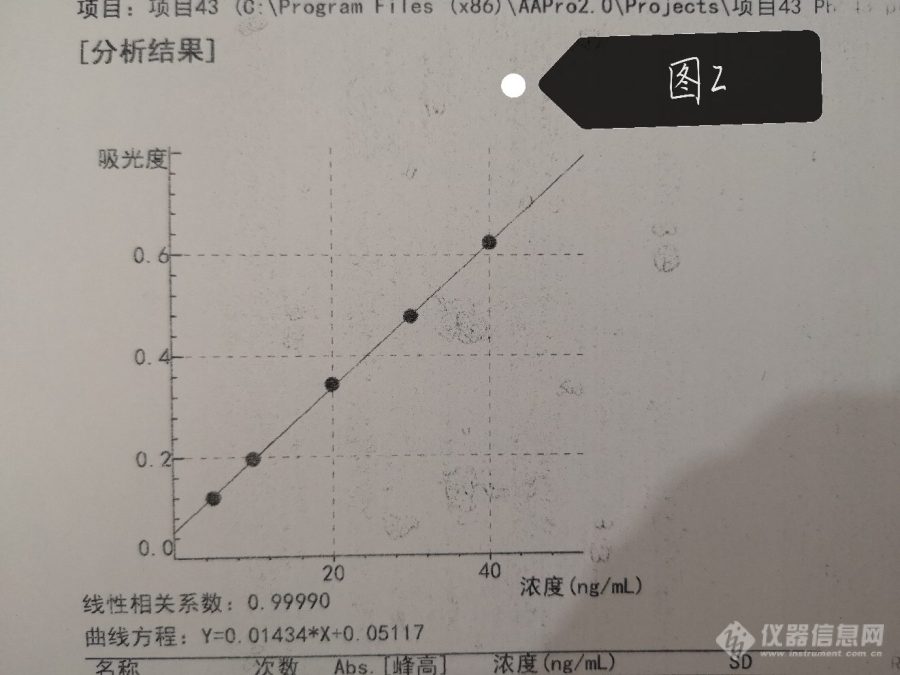
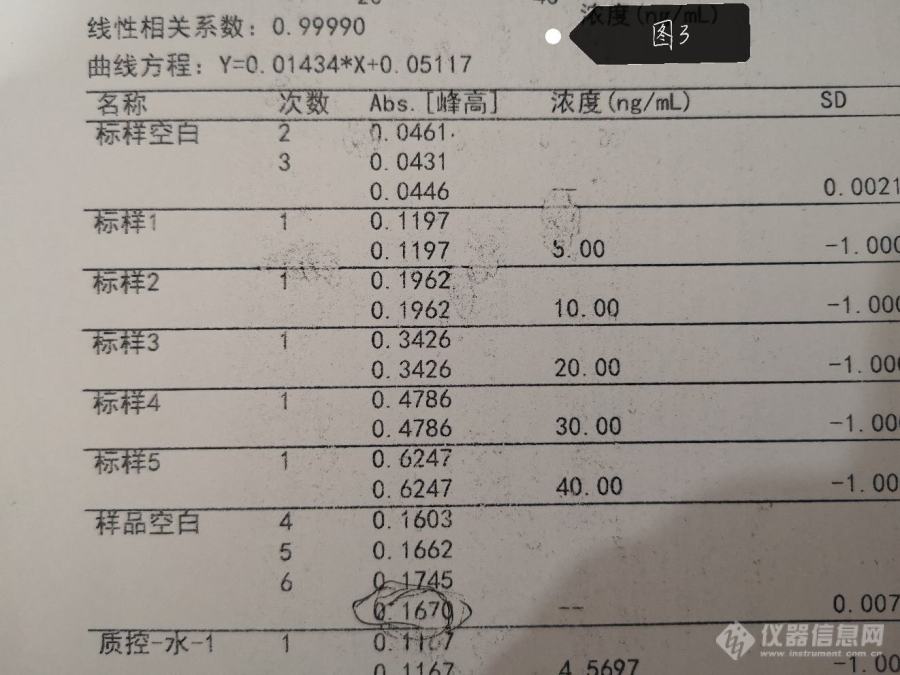
结果数据为下图2~4,标曲和样品都是去了10微升加5微升磷酸二氢铵和硝酸钯混合基改测出来的结果,还开了氘灯