-
+关注
私聊
-

第15楼2005/09/23
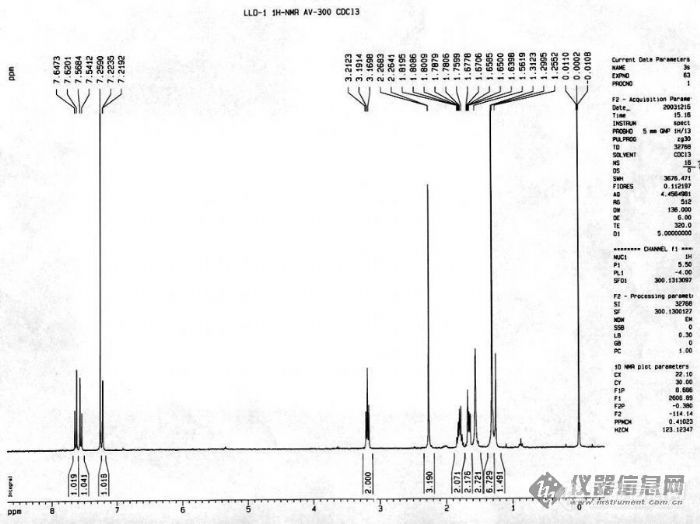
c13nmr 发表:同意sslin。CDCl3中的水峰一般在1.55附近,是比较大的宽单峰,加滴甲醇或丙酮后这个水峰会明显向低场位移。1.25ppm的峰在天然产物样品中非常常见,对应的碳峰应该是29.7ppm,这是因为样品纯化时常常有长链脂肪酸混杂在一起,(缺经验的学生尤其如此)后者的CH2信号就对应上述化学位移,如果量大点应该还可以观察到末端甲基信号(0.88ppm for proton)。非天然产物样品可能就如你说的塑料类物质(一样有长链CH2的物质)。在天然产物样品还经常看到有残留丙酮峰(2.16ppm for proton)或残留甲醇峰(about 3.5ppm for proton),这可能是样品未干或核磁管未干。
-
+关注
私聊
-
第17楼2006/09/17
花了一年的时间才搞清楚,时间太长了!象这样的问题是常见的现象,注意向有经验者请教,其实很快就可以解决.
aroar 发表:哈哈,鄙人也曾经历过此事。在我的某个样品中,曾经混有一种油状
杂质。要命的是它与我的样品比例也正好是1:1,怎么也无法搞清楚
样品的结构,差点提出一个错误的结构投了。不过,秉着科学严谨的
精神,费了我一年的功夫,最后终于怀疑是做核磁的样品混有杂质。
于是反反复复、小心的过柱子,终于把它们分开了。顺带着把杂质的
结构及来源弄明白了。不然,可就犯了一个大错误。想起来,真是不
堪回首亚。不过,吃一堑长一智嘛,现在就不会再犯这种错误了。也
算是一种收获。哈
-
+关注
私聊
-

12345890
第22楼2015/09/15
我也遇到了这样的问题,过程中也用的一次性滴管,在1.58和1.25处有峰,请问怎样将化合物提纯下,只能重新做么
will(will) 发表:你说的那个假单峰我后来发现了,的确是多重峰(少数是尖锐的真单峰,估计是其他杂质),正想告诉你呢,没想到你却已经猜到了,你真是厉害,好佩服.我的东西不是天然产品,是我自己合成的一种树枝状大分子,属于一种多肽分子.你和林老师都说得很对,的确应该是长链脂肪族化合物类的杂质,可能是硅胶里带来的或者一次性滴管带来的,或者是石油醚类的溶剂,不过在我产品里是很少很少的量了.我是想弄明白这个问题所以在这里发问了,再次谢谢各位热心回答的朋友,特别是c13nmr