2018年,美国药典(USP)通则<232>1和<233>2更新了药物、原料药和原材料中元素杂质的元素列表和每日允许暴露量(PDE)。USP现在与国际协调委员会(ICH)Q3D(R2)第5步保持一致,其最新版本于2022年通过3。
第<232>章根据给药方式介绍了元素列表及其每日允许暴露量(PDE):口服、肠外(静脉注射)和吸入药物。第<233>章详细介绍了<232>章中规定的元素分析的样品制备、分析程序和质量控制验证案。(ICH)Q3D(R2)第5步指南适用于同一组药品和原料药,并增加了皮肤和透皮给药途径的元素杂质的限度。这是一项基于风险的检测方案,用于评估是否有可能存在有毒元素。
USP<233>建议ICP-OES和ICP-MS技术均适用于杂质分析。然而,技术的选择取决于每日允许暴露量(PDE)、剂型和每日剂量。ICP-OES具有良好的灵敏度和线性动态范围,可用于部分口服药品的检测;然而,如需获得最低的检测限和最宽的校准线性范围,ICP-MS则是理想的技术。
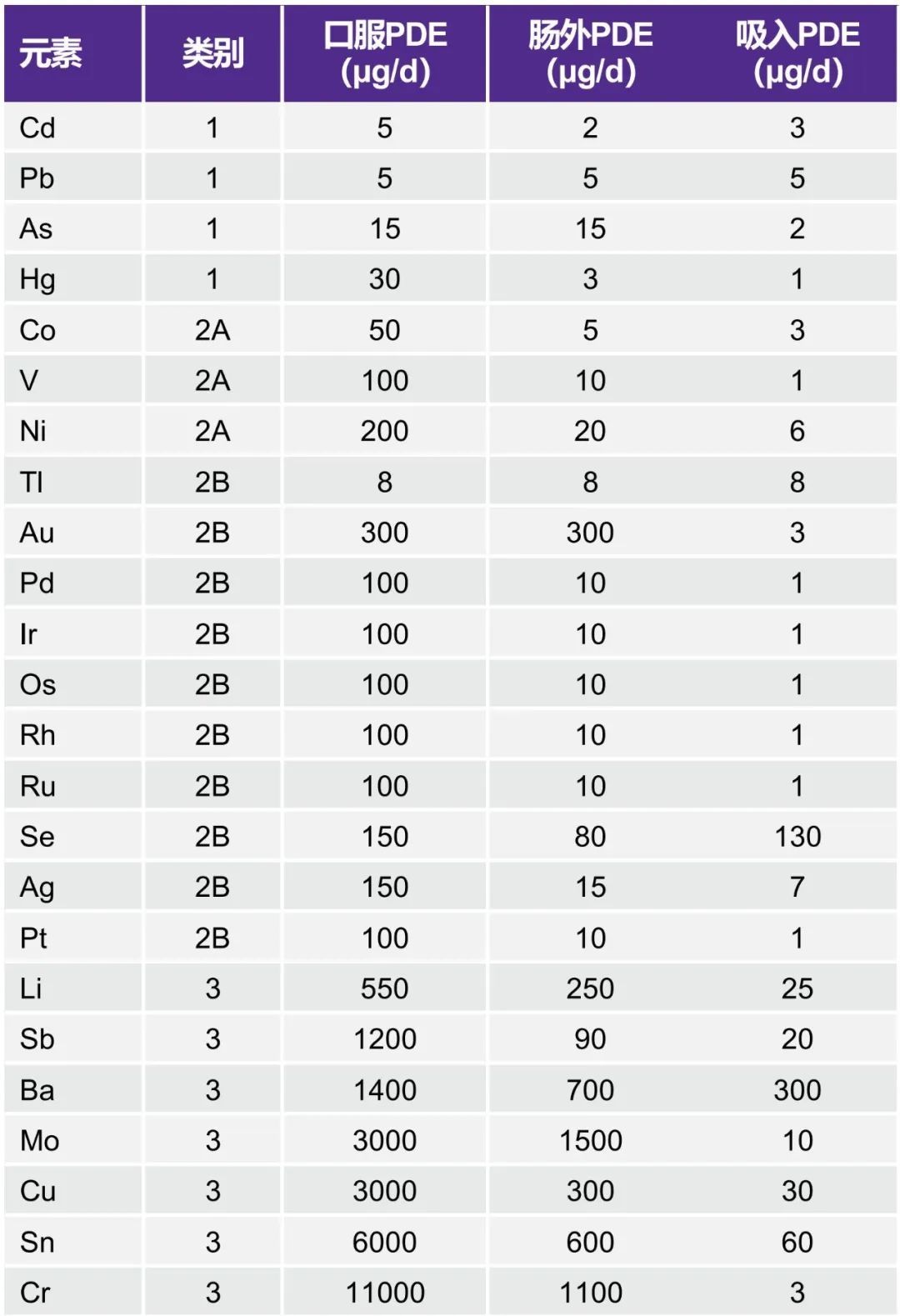
表1根据给药途径显示了ICH Q3D中的监管元素和PDE限值。根据毒性及其在药品中存在的可能性,这些元素被分为三类(1、2A/2B和3)。例如,所有类别药品的风险评估中必须至少包含1类和2A类元素。
表1. 统一的元素列表和已建立的元素杂质PDE
当PDE值(以μg/d表示)根据最大日剂量转换为μg/g或μg/L时,则称为目标限值或允许浓度。该限值在考虑稀释倍数后,称为J值,计算公式如下:

珀金埃尔默提供USP/ICH套件4,除其他组件外,还包括J计算器,用于精确计算元素杂质的J值,使标准品制备和方法开发变得更加容易。
本研究重点关注NexION® 1100 ICP-MS5在测定大容量肠外(LVP)药品中一组毒理学相关元素方面的实际效益。它概述了USP方法,特别强调杂质含量和推荐的分析程序。根据该方法的USP验证方案,给出了该系统的品质指数。
1
样品制备
用2% HNO3/2% HCl混合溶液将高纯度NaCl溶液(seaBlank,10-11% NaCl,Elemental Scientific Inc.,Omaha,Nebraska,USA)稀释11倍,制备模拟大容量肠外(LVP)生理盐水(0.9% NaCl)。将制备的模拟LVP生理盐水再用2% HNO3和2% HCl混合溶液稀释2倍至最终盐度为0.45%,然后进行分析。
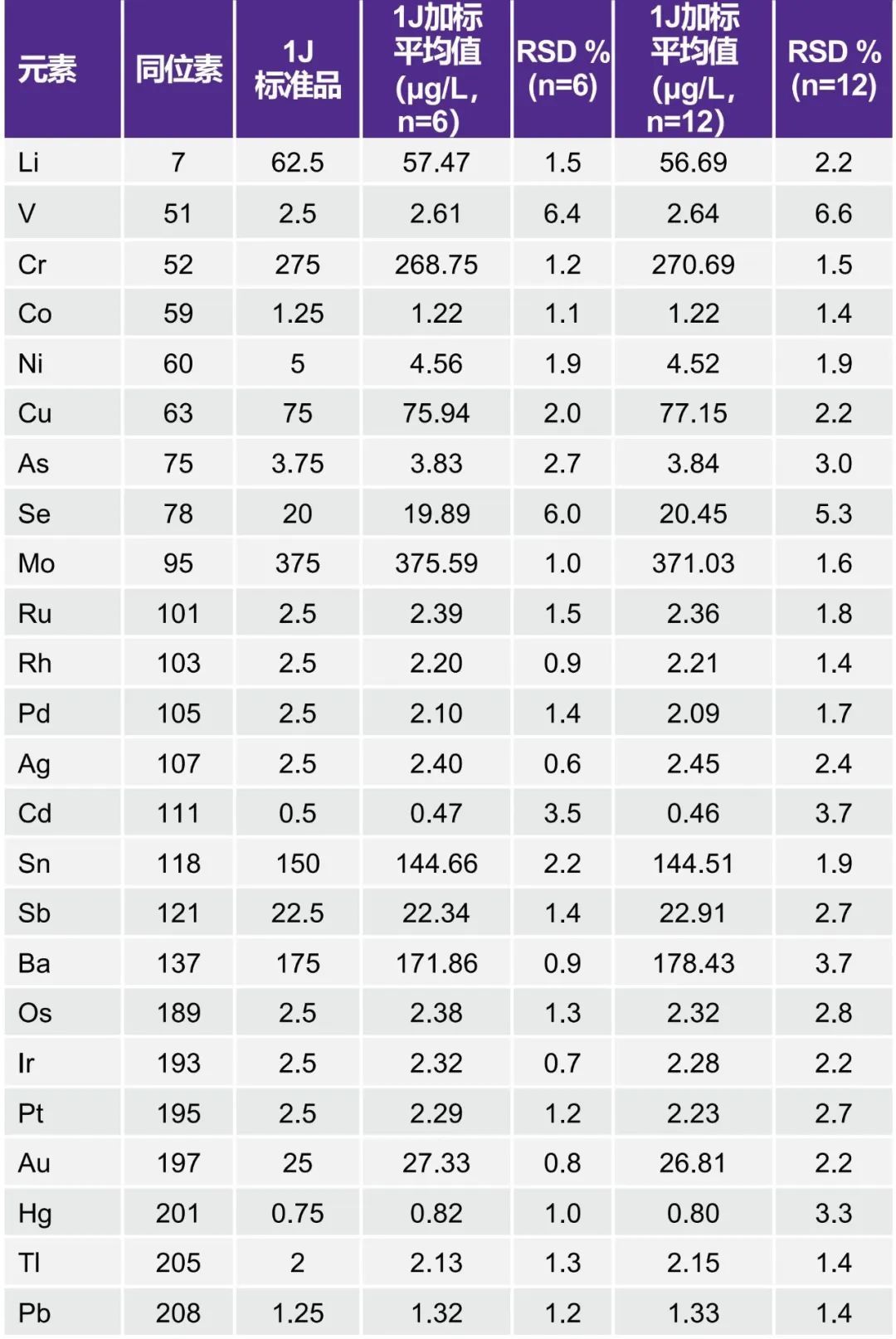
USP要求仪器校准至少基于两种标准(0.5J和1.5J)。制药公司通常使用0.3J作为风险评估。在极端情况下,如果样品含有等量的J,则添加1.5J的样品将得到2.5J的浓度。因此,低标准包括0.3J,高标准达到3J。校准标准品浓度如表2所示。校准标准品是通过将商用多元素和单元素标准品(见“所用耗材”表)稀释至基于PDE的浓度进行制备的,稀释倍数为2,生理盐水样品的剂量为2L/d。内标由1000 ppb Ge、20 ppb Tb和20ppb Bi的2% HNO3/2% HCl溶液组成,与空白、标准品和样品在线混合。
表2. LVP生理盐水的PDE(μg/d)和校准标准品浓度(μg/L)
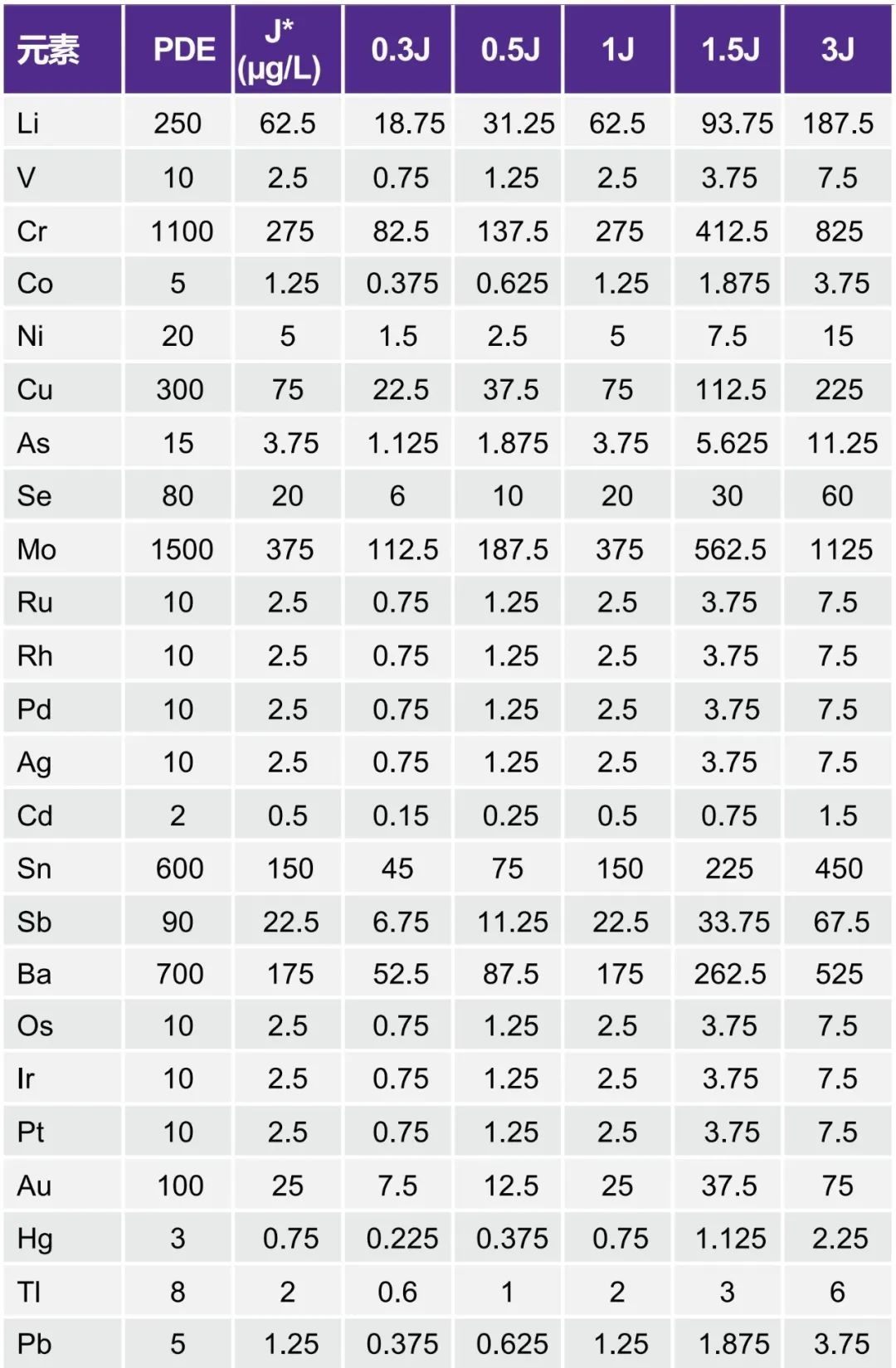
* 稀释2倍的肠外生理盐水(用于分析)
仪器
根据USP第<232>/<233>章和ICH Q3D,使用NexION 1100 ICP-MS(珀金埃尔默公司,美国康涅狄格州谢尔顿)对LVP生理盐水样品进行分析。NexION采用获得专利的离子光学设计(三锥接口-四极杆离子偏转器组合和四极杆通用池),将传统碰撞池的简便性与卓越的检测限结合在一起。在此应用中,使用氦气作为碰撞气体,使用碰撞(KED)模式测量所有元素。
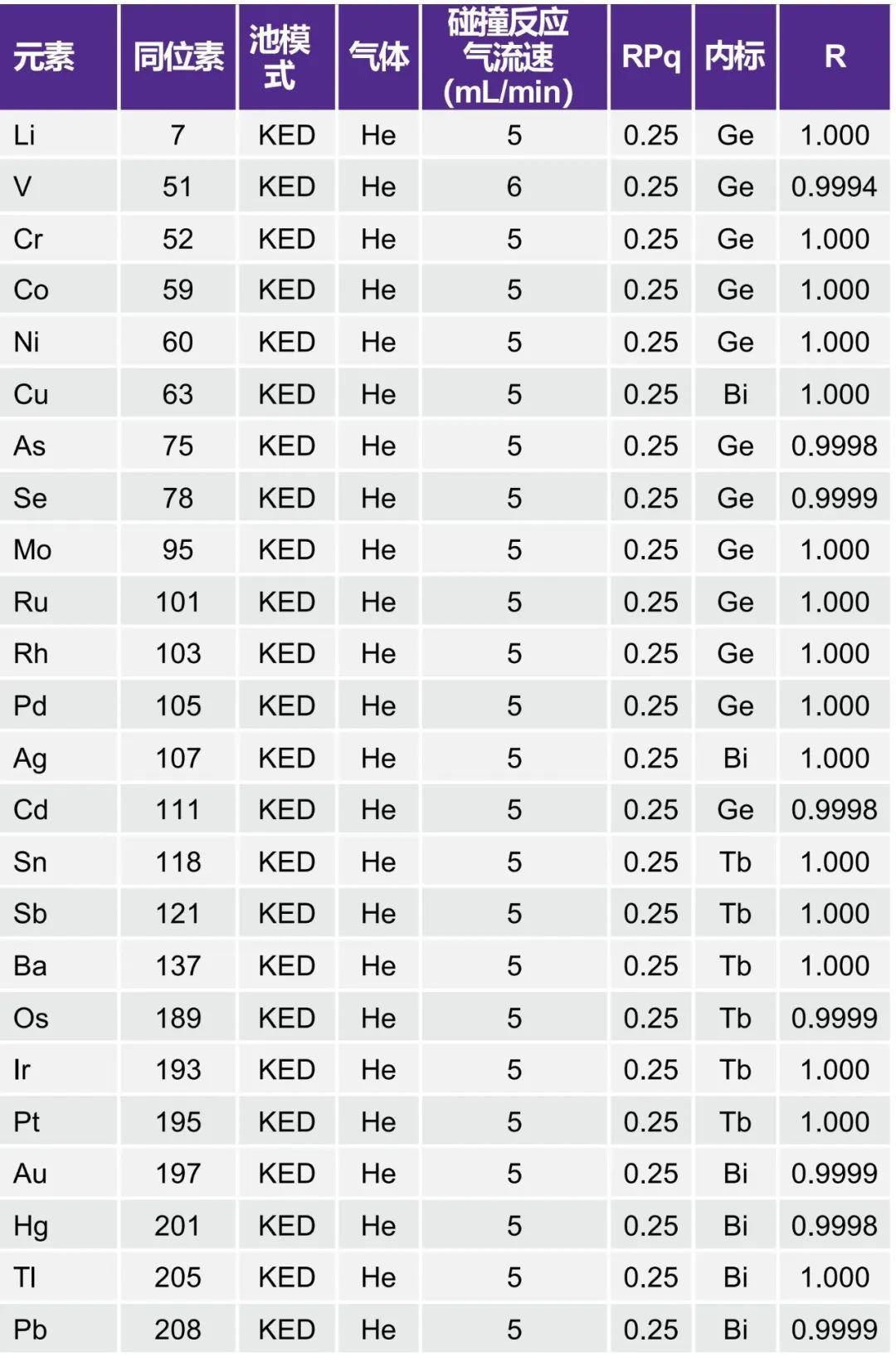
按照仪器操作手册,优化池气流以获得最佳检测限6。所有分析物均使用默认的RPq(池排斥参数),以简化方法开发。表3列出了用于分析的元素、同位素和氦(He)气流。
表3. 方法建立与线性关系
通常用于高TDS样品的全基体进样系统(AMS)均采用氩气流入喷雾室颈部以稳定信号,并提供额外的2倍稀释。
仪器工作参数如表4所示。在碰撞(KED)模式下,35Cl16O/59Co的比率通常为0.5%。对于LVP生理盐水样品的V分析,35Cl16O+/59Co+应调整至小于0.3%。在样品分析之前,需要对新清洁的锥体进行调节。该方法通过在2% HNO3和2% HCl中吸入0.45% NaCl来调节锥体,并监测内标,直至信号稳定。然后,在开始样品运行之前,先抽吸超纯水,再抽吸2% HNO3和2% HCl清洗系统。
表4. LVP生理盐水的仪器参数
2
USP<730>7和<233>定义了多个QA/QC方案来检测和验证方法开发。这些设置包括:
线性度
检测限(可检测性)
系统适用性(稳定性)
准确度
精度(重复性)
线性度
对于I类检测,相关系数的验证标准应大于0.995;对于II类定量检测,相关系数的验证标准应大于0.99。表3显示在校准范围内所有分析物的相关系数均大于0.999。
检测限
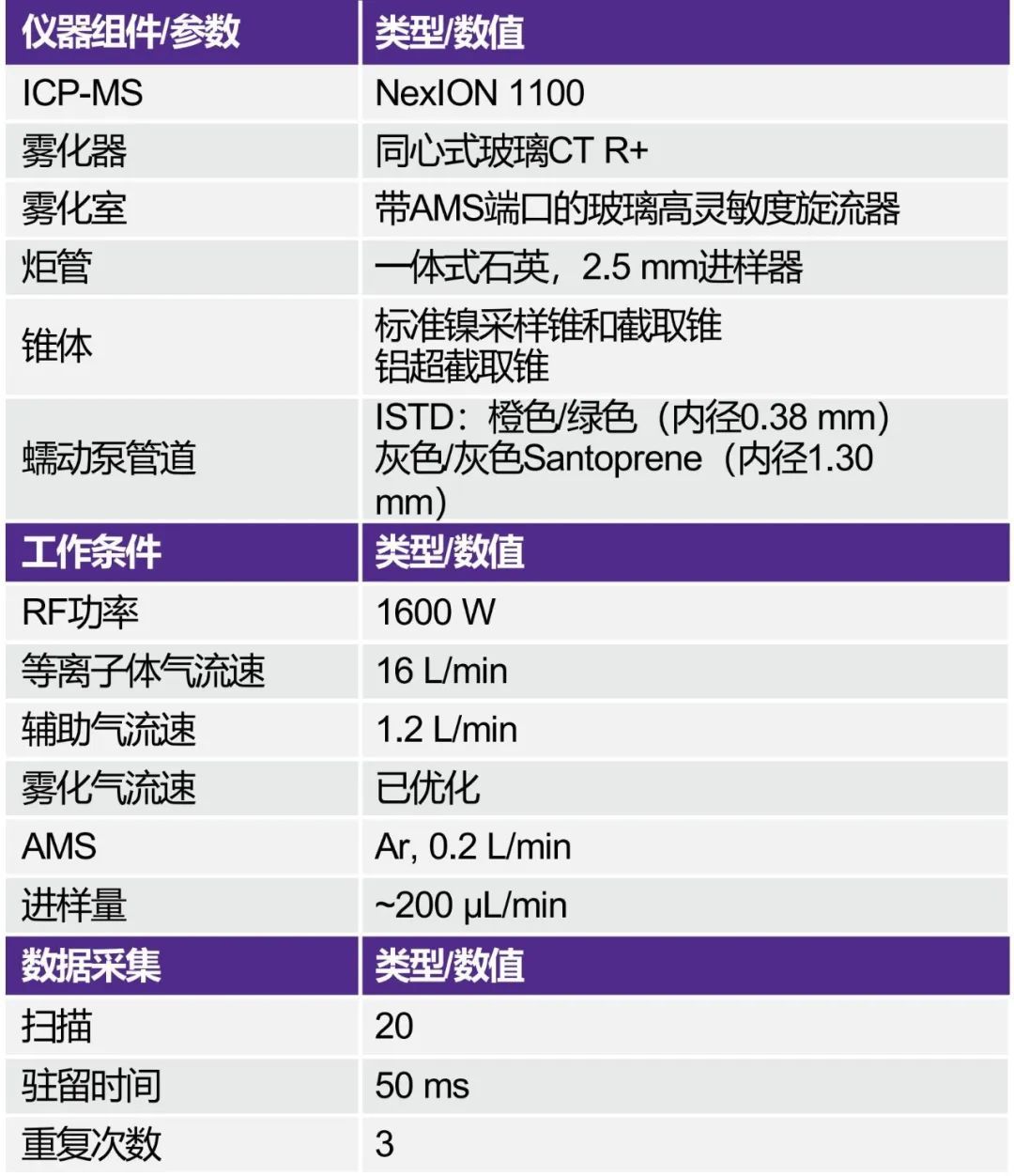
本研究中为所有元素建立了方法检测限(DL)和方法定量限(MQL),以确保方法的检测能力大大低于最大元素污染物水平(1.0J)。MDL计算为高纯度3*0.45%NaCl(n=7)三次重复测量的标准差(SD),MQL计算为10*高纯度0.45% NaCl三次重复测量的SD。结果见表5。
表5. MDL和MQL与J的比较
可检测性
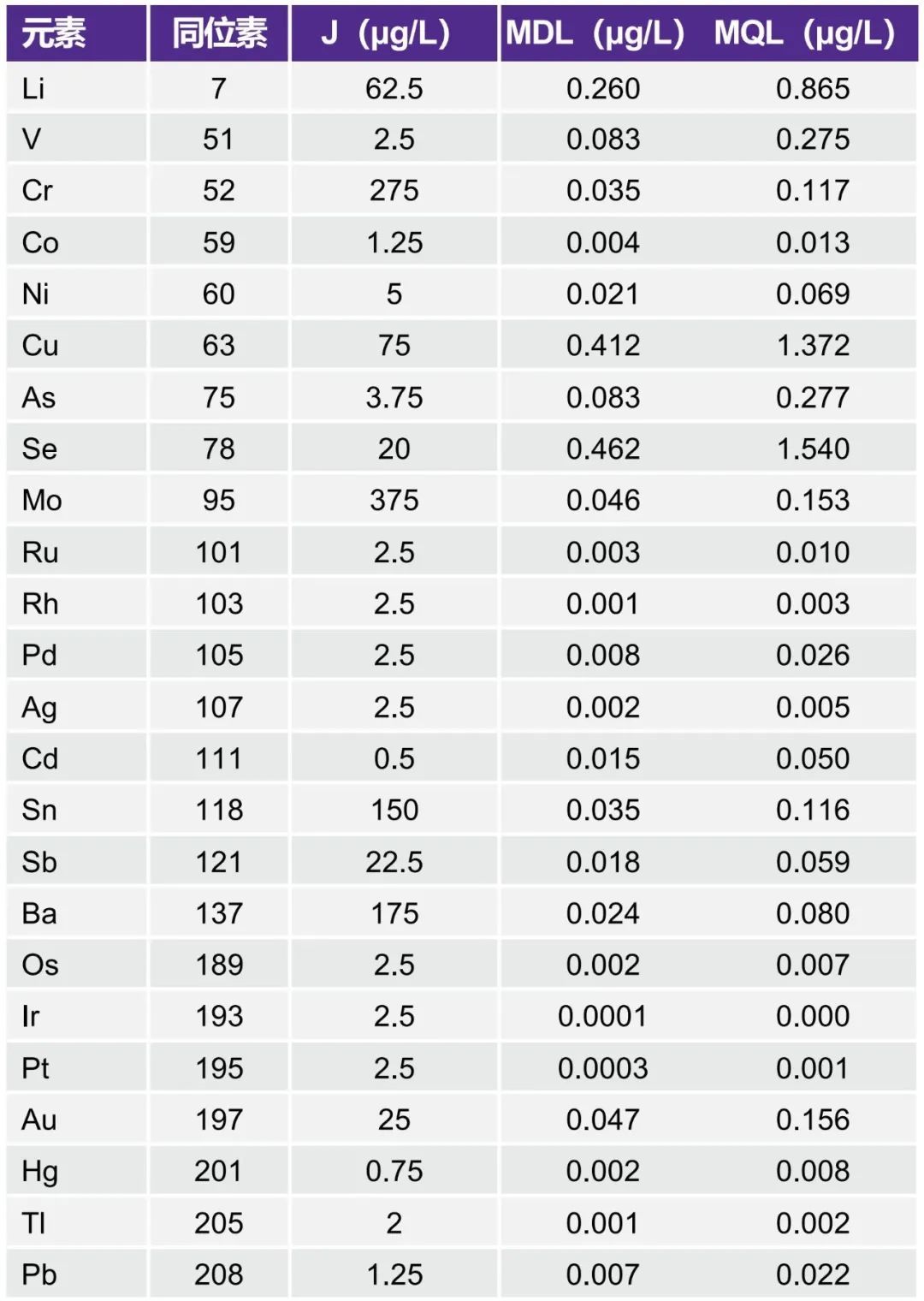
本检测对一式三份的1.0J和0.8J加标样品的平均浓度进行了比较。根据可接受标准,1.0J加标样品的平均浓度(n=3)必须在1J标准溶液的±15%范围内。0.8J加标样品的的信号强度或值必小于1J标准品。表6中的结果表明满足/超过了这些标准。
表6. 可检测性测试结果
(点击查看大图)
系统适用性(稳定性/漂移)
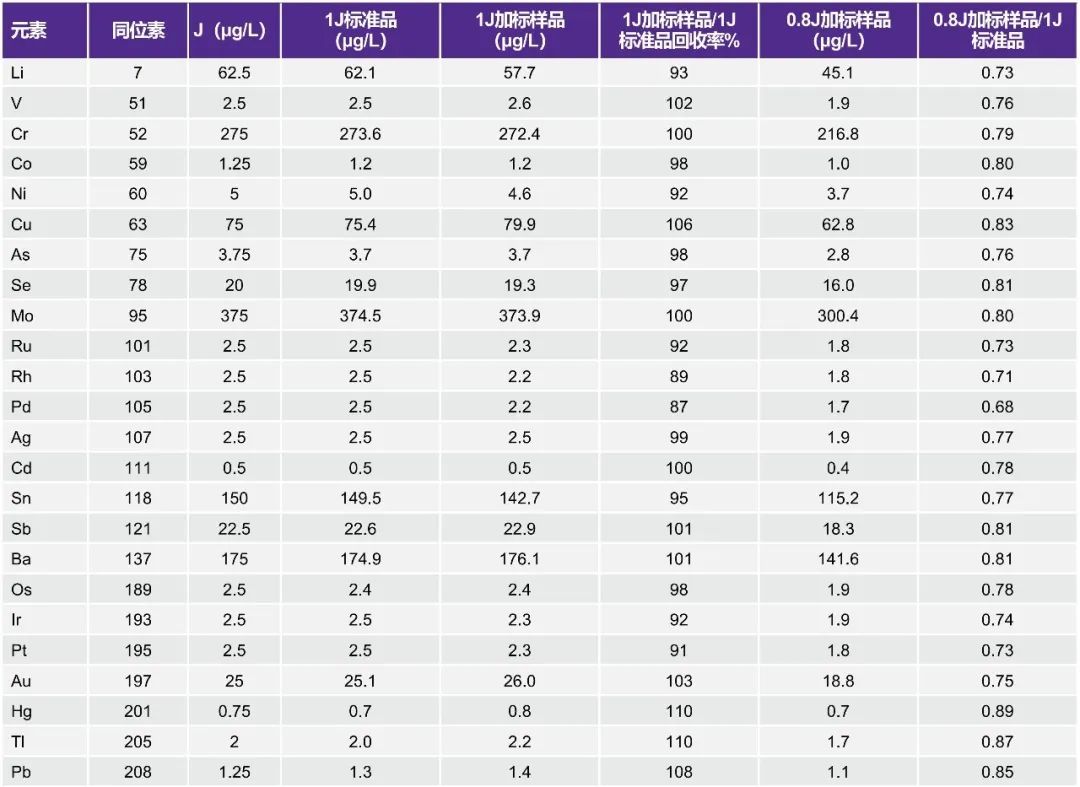
本检测需要对一批样品分析前后1.5J标准溶液的结果进行比较。可接受标准要求每个目标元素的漂移不超过20%。表7显示了在45个生理盐水样品批量运行(约2小时)前后测量的1.5J标准溶液的结果。漂移小于6.3%,很容易满足该标准。
表7. 稳定性/漂移测试结果
(点击查看大图)
准确度
采用基质加标回收率进行准确性验证。根据USP<233>,应测量浓度在0.5J ~ 1.5J之间的适当标准品以及相应的加标样品,加标回收率的可接受标准为70% ~ 150%。
然而,制药公司通常采用更严格的可接受标准,即通过评估较低水平的加标样品来获得更好的风险评估。因此,本测试测量了0.3J、1.0J和1.5J三个水平下的加标回收率。结果见表8。所有加标水平(包括0.3J加标样品)的加标回收率均在85% ~ 110%范围内。对于所有目标元素,0.3J加标样品的相对标准差(RSD,n=3)小于10%,1.0J和1.5J加标样品的RSD小于5%。
表8. 准确性评估
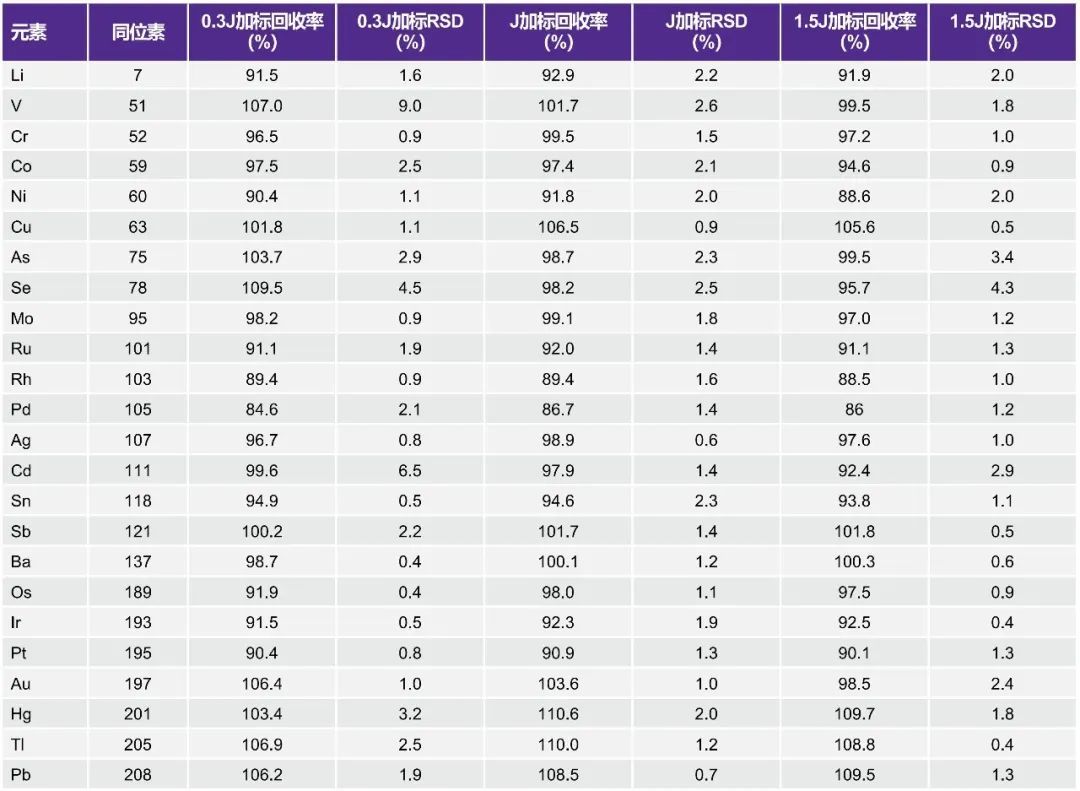
(点击查看大图)
精密度/可重复性和中间精密度
在精密度/可重复性测试中,6份1J(目标水平)加标样品中的每种目标元素的RSD应不超过20%。中间精密度测试的合格标准相同;然而,这六份样品的分析必须在不同的日期、使用不同的仪器或由不同的分析人员进行。RSD的标准等于或小于25%。如表9所示,精密度和中间精密度RSD分别<6.4%和<6.6%,远低于规格参数。
表9. 精密度/重复性和中间精密度测试结果

本研究表明,在氦碰撞(KED)模式下工作的NexION 1100 ICP-MS非常适合根据USP第<232>/<233>章和ICH Q3D进行药品分析。肠外生理盐水由于其体积大、总溶解固体(TDS)高且PDE低,是最具分析挑战的药品之一。采用氦碰撞(KED)模式对所有24种元素进行鉴定和定量分析,消除了潜在的多原子干扰,并且在线性度、检测限(可检测性)、系统适用性(稳定性)、准确性和精密度(可重复性和中间精密度)等方面均满足验证要求。
所用耗材
第A部分:仪器组件
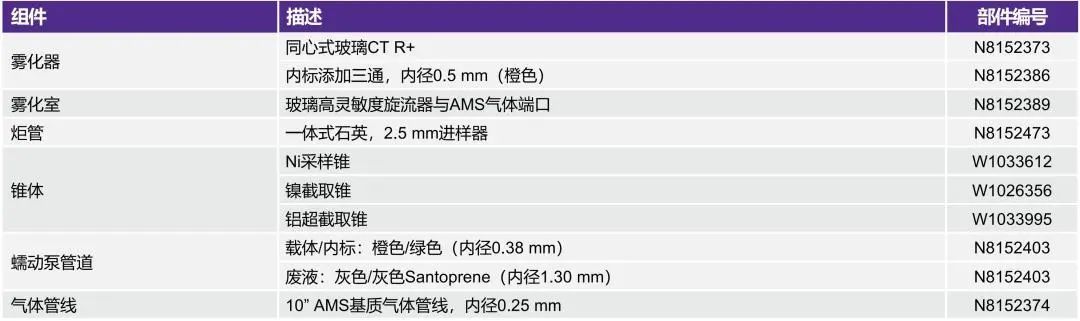
(点击查看大图)
第B部分:储备标准品

(点击查看大图)
向上滑动阅览
关注我们

Avio 200 ICP-OES测定固态电解质中杂质元素含量

与科学,共赴使命 I 珀金埃尔默与您相约analytica China 2024

Spotlight 400 红外成像系统+Spectrum Two FT-IR 优化显微红外分析微塑料的操作流程

珀金埃尔默携手ISPE(国际制药工程协会)举办Process Validation学术研讨会

相关产品
珀金埃尔默热重-红外-气相色谱质谱联用 TG-IR-GC/MS
Avio® 550 Max 珀金埃尔默高性能电感耦合 等离子体发射光谱仪
Avio 550/560 珀金埃尔默高性能电感耦合 等离子体发射光谱仪
珀金埃尔默 PerkinElmer GCMS 2400系统
珀金埃尔默GC 2400™ 平台-带分体式触摸屏 气相色谱质谱平台
珀金埃尔默 PerkinElmer MPS 320™ 微波消解仪
LAMBDA365+ 珀金埃尔默紫外/可见光谱仪
珀金埃尔默 PerkinElmer LC 300 HPLC 超高效液相
珀金埃尔默Spectrum 3™傅立叶变换红外光谱仪
珀金埃尔默 PerkinElmer PinAAcle D 900 原子吸收光谱仪
珀金埃尔默 NexION 5000 多重四极杆ICP-MS
珀金埃尔默环境监测走航系统方案
OilPrep 8 油品稀释器
OilPrep 4 油品稀释器
LPC 500液体颗粒计数器和ICP-OES油品联用系统

关注
拨打电话

留言咨询