一、法规
《药品生产质量管理规范(2010年修订)》主要是参照欧盟GMP并结合国内的实际情况进行修订的,共14章,313条,同时还新修订发布了无菌药品、原料药、生物制品、血液制品和中药制剂的附录,各项要求更加细化,明显提升了无菌制剂的洁净度要求。新版药品GMP提出了洁净区A、B、C、D分级以及相应更为严格的环境悬浮粒子与微生物的控制要求,其目的是为了防止生产中污染、混淆、交叉污染和人为差错的产生。新版GMP的发布和实施缩小了中国和国际GMP管理要求的差距,它对于推动中国制药行业的国际化进程,推动制药行业提高质量管理水平,保证人民群众用药安全将起到重要作用。
洁净室为了达到相应的室内环境,除了控制洁净室内空气的含尘度外,对细菌和微生物的存在数量也要进行控制。进入洁净室内的空气,虽然经过初效、中效、高效过滤器三级过滤,把大多数尘埃粒子及附着其上的细菌和微生物阻滞在洁净室外,但是它们仍然存在于空调系统的设备、管道、风口及过滤器上,一旦有适宜的环境就会繁衍生殖。还有少量的细菌和微生物穿透各级过滤器进入洁净室内,成为洁净室内微生物。如果不对洁净室进行消毒灭菌,它们将不断繁殖,破坏室内空气环境品质,影响产品质量。因此,应定期对洁净室进行灭菌消毒。
新版GMP无菌药品附录1第四十五条认为:必要时,可采用熏蒸的方法降低洁净区内卫生死角的微生物污染,应当验证熏蒸剂的残留水平。这也就对新版GMP下洁净厂房的灭菌设计提出了较高的要求。医药工业的洁净室传统消毒(灭菌)方式主要有:消毒液(甲醛熏蒸)灭菌法、臭氧消毒(灭菌)法。
对洁净室气体消毒的传统办法是将消毒液通过蒸发来熏蒸,通常的消毒液有环氧乙烷、过氧乙酸、甲醛溶液等。甲醛杀菌效果的3个重要因素:提升温度、限定相对湿度和改进穿透条件。当相对湿度在65%以上,温度在24~40°C时,消毒效果最好。
但甲醛熏蒸存在着一系列不可忽视的缺点:
由于每次化学试剂熏蒸消毒灭菌时间最少需24 h,不能天天进行,因此目前各生产厂家大多采用每月1次大消毒,而微生物污染源—— 人、物、料、室外新鲜空气等却天天要进出洁净室,其带入的微生物不能得到及时杀灭,会出现微生物随时间延长而增加的现象。
甲醛有强烈的刺激性气味,消毒灭菌后去除残留气味时间长,对生产操作人员身体有害。甲醛作为较高毒性的物质,具有强烈的致癌和促进癌变作用,是公认的变态反应源,也是潜在的强致突变物之一。所以,应用受到限制,欧盟GMP明确规定不能使用甲醛进行洁净厂房灭菌。
甲醛熏蒸会出现多聚甲醛聚合物(白色粉末),附着在洁净室内的维护结构和设备管道表面上,在消毒后几天内,其悬浮离子数会增加,甲醛聚合物也逐渐解聚成甲醛,对生产操作人员的危害很大。
甲醛气体的穿透性差,一般不能穿透到物品包的中心或导管的内部,不易精确控制操作过程中气体浓度,也不能保证影响甲醛杀菌能力的因素得到有效的控制。
臭氧消毒的浓度按臭氧消毒的效率和卫生部颁布的《消毒技术规范(2002年版)》的标准,对空气中的浮游菌,臭氧灭菌的浓度为2~4ppm,只需要将臭氧发生器开启1~1.5h;对物体表面的沉降菌,臭氧灭菌的浓度为10~15ppm,需要将臭氧发生器开启时间调到2~2.5h。环境条件:环境温度16~28℃;相对湿度45%~65%;室内无外界强气流,无强烈阳光照射或其他热辐射。消毒时关闭新风口和回风口排放阀门,使整个被消毒的洁净区空气通过净化系统风管形成循环,臭氧发生器开始工作。
臭氧消毒的缺点也非常明显,即高浓度对人体有害,与设备表面和建筑材料的兼容性较差,容易使铜片出现锈斑,橡胶老化,且其灭菌环境要求相对湿度>60%等。
汽化过氧化氢(VHP)灭菌技术,是利用过氧化氢在常温下气体状态比液体状态更具杀孢子能力的优点,经生成游离的羟基,用于进攻细胞成分,包括脂类、蛋白质和DNA,达到完全灭菌要求的一种技术。常用于隔离室、隔离器等密闭空间的灭菌。汽化过氧化氢(VHP)灭菌干燥、作用快速、无毒无残留,物质相容性较好,包括很多金属和塑料,适用于房间、生物安全柜、传递窗、动物笼交换站、隔离器和医疗器械等表面的灭菌消毒。生物净化时间短,根据待处理产品的物理特性,生物灭菌时间30~90min,对更广范围的微生物有效,生物灭菌循环中不产生有毒残留物,对于其他物品影响不大(装置、电器、洁净室墙板等)灭菌所需时间短,容易验证。缺点是汽化过氧化氢要做到完全汽化较难,完全做到的汽化过氧化氢造价往往比较昂贵。
德国PEA的VHP汽化过氧化氢发生器采用最新的第三代干法闪蒸技术,不仅闪蒸温度控制稳定,汽化完全,产生的过氧化氢气体粒径小,而且在灭菌的不同阶段可以设置不同的注入速率,在保证灭菌效果(可将SAL控制在10-6以下)前提下,提升物料兼容性;相对于国产设备的闭环终端喷放过氧化氢气体,PEA VHP汽化过氧化氢发生器采用开环开放式气体循环方式,气体分布系统的配合使用更使过氧化氢气体分布均匀,有效扩展消毒灭菌体积,降解速度也得到了较大的提升。而移动式脚轮设计,使得灭菌方式更加灵活,可以对不同的区域独立灭菌,相对于固定式空调机组VHP灭菌方式,无疑大大降低了成本和造价。
二、验证
验证是中外制药企业GMP管理的重要组成内容。验证的定义最早见于美国食品药品监督管理局FDA公布的《药品工艺检查验收标准》(1978年6月):“一个已验证的工艺系指已能证实按预计或所声称的那样运行的工艺。验证的证据是通过尽可能收集和评估工艺开发阶段的数据,以及以后生产阶段的数据获得的。验证必须包括工艺确认(材料、设备、系统、建筑及人员的确认),以及重复性生产的批或运行的整个工艺的控制”。
我国新颁布的《药品生产质量管理规范(2010年修订)》在第十四章第三百一十二条将验证定义为“证明任何操作规程(或方法)、生产工艺或系统能够达到预期结果的一系列活动”。GMP中验证概念的引入标志着质量管理从“质量检验”提升至“质量保证”,被称为是GMP发展史的里程碑。
无菌药品洁净区域的化学气体熏蒸灭菌,要求其可安全地使用于不锈钢、钢、塑料、玻璃、环氧地面、墙壁等各种表面,并对包括孢子在内的微生物进行快速、有效的灭菌控制,能通过对生物指示剂挑战性试验:经含菌量为105~106和103的菌条灭菌后,应能够降低相应的对数单位。
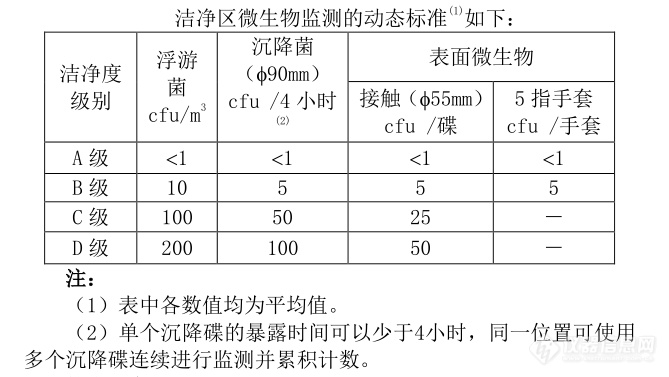
消毒灭菌方法的验证是极其重要的,只有通过验证,才能肯定消毒灭菌方式的有效性和安全性。无菌药品的要求采用了最新的世界卫生组织和欧盟 A、B、C、D 分类,对无菌药品生产的洁净度、级别提出了具体要求,要求达到动态A级的标准,增加了在线监测,特别是对悬浮粒子的静态、动态监测,对浮游菌、沉降菌、生产环境中的微生物和表面微生物的监测都作了详细的规定。
目前汽化过氧化氢灭菌法已成为各国药典、药品生产质量管理规范(GMP)、消毒灭菌技
术规范所推荐的方法,灭菌工艺已经非常成熟,重复性好,有专门的商业化的非常标准的化学指示剂和生物指示剂用来检测灭菌过程中过氧化氢气体分布的均匀性和最终的灭菌效果。
礼遇开学季 | 物美价更美?买一送一?你想要的,Countstar都给!

新技术来了 | 新一代细胞灌流系统

好用+大促=?当然是“把TA带回实验室”

艾力特生物科技2023年度全国经销商大会顺利召开







相关产品
德国PEA MLT 19ii气化过氧化氢空间灭菌器
德国MMM集团Unisteri脉动真空压力蒸汽灭菌柜
德国MMM Venticell强制对流烘箱
德国PEA汽化过氧化氢传递窗
罗氏Cedex Bio与Cedex Bio HT分析仪
德国PEA VHP气化过氧化氢灭菌器
Countstar BioFerm自动酵母计数仪
Countstar BioMed 自动细胞计数仪
Countstar BioMarine 自动藻类计数仪
Countstar BioLab自动细胞计数仪
Countstar BioTech 自动细胞计数仪
Countstar Rigel 全自动细胞荧光分析仪
Countstar AItair 全自动细胞分析仪
Countstar Mira BF 全自动细胞分析仪
VHP汽化过氧化氢灭菌器

关注
拨打电话

留言咨询





