
方案摘要
方案下载| 应用领域 | 环保 |
| 检测样本 | 其他 |
| 检测项目 | |
| 参考标准 | / |
卡尔费休滴定是水份含量测定的一种方法,随着梅特勒-托利多的新一代紧凑型容量法 和库仑法卡尔费休滴定仪V20/V30,C20/C30以及超越系列组合型容量法滴定仪T70和 T90的问世,水份含量的测定达到了无与伦比的简单和可靠。通常这是最理想的水份测 试的方法,可以帮助您轻松应对日常工作。

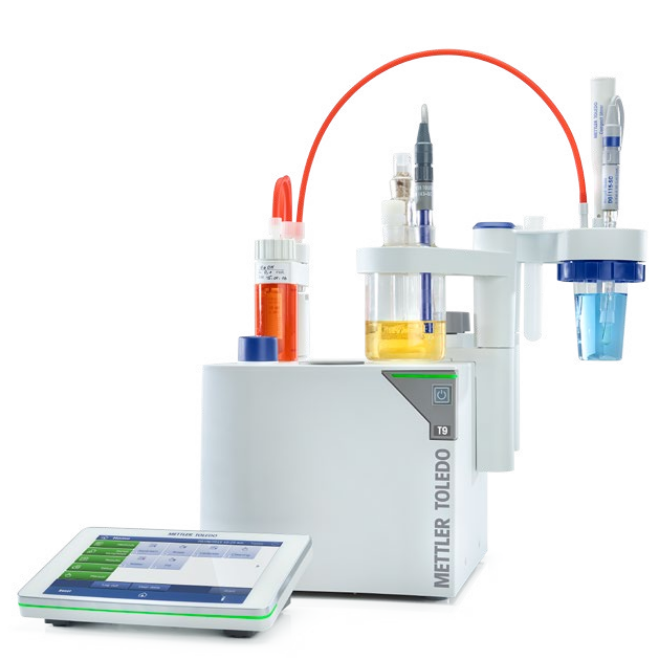
容量法和库仑法卡尔费休分析
卡尔费休水分测定有两种方法:
-容量法卡尔费休滴定,含碘的溶液通过活塞驱动的滴定管添加;
-库仑法卡尔费休分析,碘由杯中的电解电极产生
根据水分的预估量选择不同的测定方法:
容量法卡尔费休滴定
碘通过滴定管添加。
适用于较大水分含量:100ppm-100%
库仑法卡尔费休分析
碘通过电解产生。
适用于痕量水分含量:1ppm-5%
卡尔费休滴定
在卡尔费休滴定中,为了得到正确的结果,必须考虑下列因素:
空气湿度
工作环境
样品 pH 值
样品与卡尔费休试剂是否有副反应
卡尔费休库仑滴定仪的使用
库仑滴定杯的加液
有隔膜电解电极滴定杯:
-首先将5 mL 阴极液注入阴极池内。
-用注射器从瓶子取5mL阴极液注入到电解电极内,或直接将一安瓿瓶阴极液倒入电解电极内。
-然后加入约 100 mL 阳极液。
-确保阳极液的液面高于阴极液液面约3~5mm。阴极液含有痕量的水分,如果阴极池的液面等于或高于阳极池液面,液体会从阴极池扩散到阳极池,水分进入隔膜并缓慢释放至阳极池,导致比较高的漂移。可以通过以下方式避免:
1.阴极液可以加入几滴单组分卡尔费休试剂来去除里面的水分。
2.确保阳极液的液面高于阴极液的液面。
-阳极液和阴极液液面高度不同指的是在搅拌的情况下,当搅拌停止时,两种电解液液面会趋于一致。
-当样品注入阳极池内时,阳极液液面的高度增加。但是,当联用加热炉时,阳极液液面的高度会由于蒸发而降低,这时要在阳极液中不时加入无水甲醇以维持阳极液的液面高度。
无隔膜电解电极滴定杯
- 加入约 100 mL电解液(阳极液)至滴定杯中。
-电解电极浸入电解液约2.5 cm。
注意:在碘产生过程中,阳极液会存在一些过量的碘而呈棕色。通常在滴定杯中加入样品,会由于碘被消耗而使溶液的棕色消失,如果颜色没有消失,必须加入一些甲醇或者样品至滴定杯中,使溶液的颜色变为黄色。
何时更换电解液
出现下列情况必须更换电解液:
如果电解液的电解能力耗尽。
- 阳极液(100 mL):1000 mg 水以后,
- 阴极液(5mL):200 mg 水以后。
电解液的电解能力由库仑滴定仪指示(参见操作手册)。
实际上,当使用有隔膜电解电极时,阳极液和阴极液需要同时更换。
如果加入样品后,溶剂或者阳极液的液面超过滴定杯标记的150mL 时。阳极液的液面越高,搅拌效果越差,过滴定的风险增加。
如果阳极液的电导率降低至很低时,比如10μS。
如果滴定了大量的电导率低的样品后,可能会出现这种情况。
如果阳极池内形成乳浊液。
对于溶解性差的样品,阳极液(库仑法卡尔费休试剂〉的溶解能力耗尽,形成乳浊液,进而导致错误的结果。
如果漂移值太高。
如果电解液使用一段时间以后没有更换,漂移值会逐渐升高。
使用两周后
阴极池内会生成硫化物和硫醇,产生难闻的气味,导致漂移值很高。
滴定杯的安全加液和排空:溶剂管理器
库仑滴定杯加液和排空的过程中,最简单最安全的操作方式是采用外置的卡尔费休液体处理器-溶剂管理器。溶剂管理器被放置在废液瓶上,具有动力强劲的空气泵(隔膜泵)和电磁阀,完全由滴定仪控制。内置的液位传感器可以防止废液溢流,在排空和加液的过程中完全避免与试剂接触。
卡尔费休滴定杯的清洗
滴定杯、电解电极和测量电极均需清洗,特别在分析“脏”的样品之后。有隔膜电解电极必须定期清洗,因为使用一段时间后,污染物会沉积,导致漂移值升高,此污染物可能来源于样品或者阴极池中还原产生的副产物。
·滴定杯的清洗
用水或者适合的溶剂清洗滴定杯,然后在100℃的烘箱内干燥或者用热空气干燥。如果需要马上使用,可以用无水甲醇冲洗。
电解电极的简单清洗和干燥
方法A:
-将空的电解电极浸在无水甲醇中。
-甲醇通过隔膜渗入阴极池,隔膜上的水分和污染物被冲洗掉。
方法B:
-将无水甲醇加入电解电极的阴极池中,甲醇通过隔膜冲洗掉水分和污染物。
-之后至少再重复一次。
-在不超过50℃的烘箱中干燥或者用热空气干燥,如果马上使用不需要干燥。
电解电极的彻底清洗
如果电解电极和隔膜污染严重,最有效的清洗剂是含三价铬的铬酸洗液。清洗过程如上所述,将空的电解电极浸在铬酸洗液中,或者注入电解电极的阴极池,让铬酸洗液流出。
然后用水彻底清洗电解电极,用无水甲醇冲洗,并安上述方法干燥处理。
测量电极的清洗
通常测量电极不需要清洗。前面已经提到,在完成最初的几个滴定后,铂环表面会形成一层薄膜,这会导致电位突越升高,这层薄膜不能通过清洗去除。
然而样品可能会沉积在电极表面,使电极的欧姆电阻增加,进而妨碍电极的正常指示。这点也可以通过终点时阳极液颜色变深来判断,这时测量电极必须清洗。
-用纸巾擦拭电极,多数情况下这样做足够了。
-如果电极污染严重,可以将测量电极浸在0.5 mol/L硫酸溶液中,启动卡尔费休滴定,并通电 400 mA 持续 60 秒。
1) 断开测量电极的S7 接口。
2) 接通电解电极和测量电极的S7 接口。
3) 清洗结束后停止此方法,测量电极的极化电流也就停止了。
样品
取样
当取样进行水分测定时,必须尽量避免空气总的水分影响一这是导致误差的最主要因素。如果在取样时吸收或损失了水分而造成样品含水量的变化,那么将得不到准确的水分含量。
取样时,请注意下列事项:
1. 样品应具有代表性,即取的样品的水分含量和整个样品的平均水分含量相同。
2. 取样时动作必须快,或至少避免水分的损失或吸收。
3.样品中存在非均质水:
在油等非极性液体中,水分的分布不均匀,或漂在表面或沉入底部。这类液体在取样前
必须混合均匀(通过震摇)。
在黄油等不能像液体一样混合的非极性固体中,取样时多取一些可以使水分分布均匀。
4.易吸湿的固体如果在存储过程中吸收了空气中的水分,那么它表面的水含量可能高于内
部的水含量。
5. 极低水分含量的物质:
极低水分含量的物质会持续快速的吸湿,因此这类样品必须用绝对干燥的注射器或者药
匙快速取样。
存储样品
取样后必须尽快测定水分含量,如果需要存储样品,必须存放在完全密封的瓶中:
-玻璃瓶比塑料瓶更好,因为塑料不能完全绝水的,空气中的水分可能通过塑料渗入其中
使样品吸湿。
-使用小瓶口的瓶子以尽量减少水分进入。
-对于极低含水量的液体,最好使用带橡胶塞的瓶子。
-选择与取样量体积相当的瓶子:
样品上方的空间越小,存在的空气水分就越小。
-对于液体样品,瓶子最好先用少量样品润洗2~3次。
对于与水不相溶的油类样品,长期存储过程中如果样品冷却或水的溶解性下降时,会导致水分从样品中分离出来。这可以通过加入助溶剂如异丙醇来增加水的溶解性。
样品量
样品的取样量取决于:
-预计的水分含量,和
-对精确度的需求
对于库仑法分析,每次取样的最佳含水量在0.5~2 mg之间,即使每次取0.1 mg的样品也能得到重复性好的结果。在优化测定条件的情况下,如果对于重复性要求不高,大约10~50 μg 的水也可以测定。
通常,样品量越大精确度越高,因为在取样和加样过程中,吸收的空气水分相对样品水分的比重会减少。
原理:
取样量可以通过预计含水量和最佳含水量决定。
步骤:
从最佳含水量(对于容量法10 mg,对于库仑法1mg)或预计的含水量开始。
用直线连接最佳含水量和预计含水量。
直线与“样品量”的交点读数即为建议取样量。
注意:刻度为对数关系!
举例:
预计含水量:5000 ppm
最佳含水量:10 mg/个样品
最佳取样量:2g
加样
液体样品
加入液体样品时,必须注意避免样品吸收空气中的水分,特别是低含水量的样品。
从具塞样品瓶中取样
取几次样品之后,由于橡皮塞的密封,瓶中的真空度增加,就不能再取样了。为了避免出现这种情况,必须通入干燥空气(平衡气压)。
拔出注射器的活塞,填充入分子筛,用脱脂棉封住,然后将短针头插入密封的样品瓶中。当取样时,空气通过分子筛干燥就可以进入瓶中了。
液体样品用减量称重法加入
一用注射器抽取1/4 管样品,如果样品易吸湿或者含水量滴(<1000 ppm),用密封的样品瓶并平衡气压。
-抽回活塞,用样品润洗注射器并震摇。
- 排空注射器(至废液瓶),重复润洗2~3次。
- 用注射器抽取样品,用纸巾擦干针头上的液体。
-将注射器放在烧杯(针头朝上)中置于天平称盘上,并清零。
-按“开始样品”启动滴定方法。
- 将样品透过隔膜注射入滴定杯中。
-抽回活塞使针头上的液滴吸回到针头内,否则拔出注射器的时候,液滴会粘附在隔膜背面。
-将剩余样品的注射器放回天平,减量称重。
- 在滴定仪上按“样品数据”,输入样品大小或者自动传输称量数据。
- 自动滴定
固体样品
不能用卡尔费休库仑法直接滴定固体样品一由于空气湿度的不同,当打开滴定杯加样时,大
约会有 50~100μg的水会进入阳极池。对于1mg/个样品的取样量,这些水分将导致 5~10%的误差。因此用卡尔费休库仑法测定低含水量的固体样品时必须采用其他方法:
●外部萃取
●外部溶解
●干燥炉
另一方面,在容量法卡尔费休滴定仪上可以直接将固体样品加入滴定杯中。样品应该尽可能快的称量并加入,尽量避免与空气接触。如果可能,可以在与样品运输和存储相同的条件下加样另外冷藏存储的样品可能会导致水气凝结,所以在称样前必须在密闭的容器中加热至室温。
固体样品用减量称重法加入
-用称量舟称取样品。
-天平清零。
将样品加入滴定杯,如果需要可以用连接软管的称量舟,以防止样品附着在滴定杯壁或电极表面上。
-减量称重空的称量舟。
-在滴定仪上按“样品数据”,输入样品大小或自动传输称量数据。
-开始滴定。
容量法卡尔费休滴定仪
问题 | 可能产生的原因及处理方法 |
滴定溶液变黑 | 过滴定 |
新加入的溶剂、预滴定结束后,漂移值太高 | 滴定台有空气水分进入 |
待机时漂移值太高 | 滴定台有空气水分进入 |
测定一个样品之后,漂移值太高 | 样品没有完全溶剂,并持续释放水分 |
滴定时间特别长,达不到终点 | 错误的终止参数 |
重复性差 | 样品量太小 |
测量值太低 | 滴定时间过短 |
单个值太高 | 控制参数太快(=略微过滴定) |
一个样品系列的结果呈递减趋势 | 溶剂的溶解能力耗尽 |
使用双组分试剂时,滴定速度很慢 | 溶剂中的二氧化硫含量不够 |
用水标定浓度时,结果错误或数值剧烈变化 | 用水标定浓度需要练习 |
用二水合酒石酸钠标定浓度时,结果递增 | 二水合酒石酸钠的溶解性差 |
校验干燥炉超限重复性差 | 卡尔费休干燥炉标准水5.55%,在220℃,15~20分钟条件下 |
文献贡献者


梅特勒托利多关于锂电行业分析仪器解决方案

梅特勒托利多 | 食品行业水份测定方法
梅特勒 电位滴定仪在半导体行业的应用
相关产品
梅特勒ME104E分析天平
梅特勒ME204E /02分析天平
YSI ProDSS/ProDSS-C手持多参数水质测量仪
梅特勒 智能银量法滴定电极DMI141-SC
梅特勒MX303精密天平
梅特勒MR203精密天平
梅特勒MA54分析天平
赫施曼 六联固定剂箱
OI 1080 TOC 总有机碳分析仪
Hirschmann/赫施曼SOLARUS光能电子滴定器 9392050
SonTek 智能多频走航式多普勒流速剖面仪系统M9/S5型
WTW Multi 3630 IDS便携式多参数分析仪
YSI Pro20手持式野外溶解氧测量仪(膜法)
WTW DIQ/S 181单通道控制器
WTW Turb 430T便携式浊度仪

关注
拨打电话

留言咨询








