
高分子表征技术专题——示差扫描量热法进展及其在高分子表征中的应用


导读:本文从热分析基础出发,依次对传统DSC、TMDSC和FSC进行了介绍,内容覆盖其发展历史、方法原理、操作技巧及其在高分子表征中的应用举例,最后对DSC未来的发展和应用进行了展望。
2021年,《高分子学报》邀请了国内擅长各种现代表征方法的一流高分子学者领衔撰写从基本原理出发的高分子现代表征方法综述并上线了虚拟专辑。仪器信息网在获《高分子学报》副主编胡文兵老师授权后,也将上线同名专题并转载专题文章,帮助广大研究生和年轻学者了解、学习并提升高分子表征技术。在此,向胡文兵老师和组织及参与撰写的各位专家学者表示感谢。

高分子表征技术专题前言
孔子曰:“工欲善其事,必先利其器”。 我们要做好高分子的科学研究工作,掌握基本的表征方法必不可少。每一位学者在自己的学术成长历程中,都或多或少地有幸获得过学术界前辈在实验表征方法方面的宝贵指导!随着科学技术的高速发展,传统的高分子实验表征方法及其应用也取得了长足的进步。目前,中国的高分子学术论文数已经位居世界领先地位,但国内关于高分子现代表征方法方面的系统知识介绍较为缺乏。为此,《高分子学报》主编张希教授委托副主编王笃金研究员和胡文兵教授,组织系列从基本原理出发的高分子现代表征方法综述,邀请国内擅长各种现代表征方法的一流高分子学者领衔撰写。每篇综述涵盖基本原理、实验技巧和典型应用三个方面,旨在给广大研究生和年轻学者提供做好高分子表征工作所必须掌握的基础知识训练。我们的邀请获得了本领域专家学者的热情反馈和大力支持,借此机会特表感谢!
从2021年第3期开始,以上文章将陆续在《高分子学报》发表,并在网站上发布虚拟专辑,以方便大家浏览阅读。期待这一系列的现代表征方法综述能成为高分子科学知识大厦的奠基石,支撑年轻高分子学者的茁壮成长!也期待未来有更多的学术界同行一起加入到这一工作中来。
高分子表征技术的发展推动了我国高分子学科的持续进步,为提升我国高分子研究的国际地位作出了贡献. 借此虚拟专辑出版之际,让我们表达对高分子物理和表征学界的老一辈科学家的崇高敬意!


原文链接:
http://www.gfzxb.org/article/doi/10.11777/j.issn1000-3304.2020.20234
《高分子学报》高分子表征技术专题链接:
http://www.gfzxb.org/article/doi/10.11777/j.issn1000-3304
示差扫描量热法进展及其在高分子表征中的应用
陈咏萱 , 周东山 , 胡文兵
南京大学化学化工学院 配位化学国家重点实验室机构 南京 210023
作者简介: 胡文兵,男,1966年生. 南京大学化学化工学院高分子系教授、博士生导师. 1989年本科毕业于复旦大学材料科学系,1995年博士毕业于复旦大学高分子科学系. 分别于1998~1999年赴德国弗莱堡大学物理系、2000~2001年美国田纳西大学化学系、2001~2003年荷兰物质科学研究院(FOM)原子与分子物理研究所从事博士后研究. 2004年至今,在南京大学任教. 2008年获杰出青年科学基金资助,2020年入选美国物理学会会士(APS Fellow). 主要研究方向为采用蒙特卡洛分子模拟和Flash DSC研究高分子结晶机理及材料热导率表征;
通讯作者: 胡文兵, E-mail: wbhu@nju.edu.cn
摘要: 示差扫描量热法(DSC)是表征材料热性能和热反应的一种高效研究工具,具有操作简便、应用广泛、测量值物理意义明确等优点. 近年来DSC技术的发展大大拓展了高分子材料表征的测试范围,促进了对高分子物理转变的热力学和动力学的深入研究. 温度调制示差扫描量热法(TMDSC)是DSC在20世纪90年代的标志性进展,它在传统DSC的线性升温速率的基础之上引入了调制速率,从而可将总热流信号分解为可逆信号和不可逆信号两部分,并能测量准等温过程的可逆热容. 闪速示差扫描量热法(FSC)是DSC技术近年来的创新性发展,它采用体积微小的氮化硅薄膜芯片传感器替代传统DSC的坩埚作为试样容器和控温系统,实现了超快速的升降温扫描速率以及微米尺度上的样品测试,使得对于高分子在扫描过程中的结构重组机制的分析以及对实际的生产加工条件的直接模拟成为可能. 本文从热分析基础出发,依次对传统DSC、TMDSC和FSC进行了介绍,内容覆盖其发展历史、方法原理、操作技巧及其在高分子表征中的应用举例,最后对DSC未来的发展和应用进行了展望. 本文希望通过综述DSC原理、实验技巧和应用进展,帮助读者加深对DSC这一常用表征技术的理解,进一步拓展DSC表征高分子材料的应用.
关键词: 高分子表征 / 示差扫描量热法 / 温度调制示差扫描量热法 / 闪速示差扫描量热法
目录
1. 热分析基础
1.1 温度和热
1.2 热分析(thermal analysis)
2. 示差扫描量热法
2.1 基本原理
2.2 实验技巧
2.2.1 仪器校准
2.2.2 样品制备
2.2.3 温度程序
2.2.4 保护气氛
2.3 应用举例
2.3.1 比热容
2.3.2 热转变温度
2.3.3 转变焓
2.3.4 DSC与其他技术连用
3. 温度调制示差扫描量热法
3.1 基本原理
3.2 实验技巧
3.2.1 样品质量
3.2.2 温度程序
3.3 应用举例
3.3.1 可逆热容和不可逆热容
3.3.2 等温可逆热容
3.3.3 玻璃化转变
4. 闪速示差扫描量热法
4.1 基本原理
4.2 实验技巧
4.2.1 样品制备
4.2.2 样品质量
4.2.3 临界条件
4.3 应用举例
4.3.1 等温总结晶动力学
4.3.2 不可逆熔融转变
4.3.3 与其他表征技术连用
4.3.4 玻璃化转变
4.3.5 热导率
5. 总结与展望
参考文献
1. 热分析基础
1.1 温度和热
温度是表征物体冷热程度的物理量,它仅由系统内部的热运动状态决定,是系统中物质分子热运动强度的量度. 热力学第零定律表明,所有互为热平衡的系统都存在一个共同的数值相同的态函数,这个态函数被称为温度,是一个强度量. 热力学第零定律阐明了温度计的工作原理:在测量温度时,首先选择一个作为标准的测温物体,也就是温度计,然后让它分别与各个物体接触并达到热平衡,得到的标准物体的温度就是各待测物体的温度. 值得注意的是,温度计的热容必须比待测物体的热容要低得多,以保证接触过程中不会改变物体的温度. 然而,温度测量获得的是一个相对量,为了定量测定温度,人们还需要建立一个温标.
最初的温标是经验温标,它依据测温质的某一种物理属性随温度的变化关系来表征温度的大小. 例如,酒精和水银温度计是根据液体加热时的体积膨胀设计的,铂和RuO2温度传感器是依据金属导体的电阻随温度的变化关系设计的. 通常,这种变化关系是显著而单调的,假定其为简单的线性关系,那么测温属性x和温度θ的关系为:
|
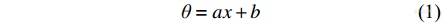
其中,常数a和b是由标准点和分度法确定的,根据不同的标准点和分度法可以确定不同的温标. 1714年,Fahrenheit将水的冰点设为32 °F,沸点为212 °F,建立了华氏温度. 1742年,Celsius将水的冰点设为0 °C,沸点为100 °C,建立了摄氏温度. 到1779年为止,全世界并存有19种经验温标. 然而,这些温标缺乏统一的标准,除了标准点外,采用不同的测温质测得的温度并不完全一致. 此外,测温属性往往无法在整个温度范围内保持完全线性的变化关系. 例如,水银在−39 °C发生固化,在357 °C发生气化,因此水银温度计的测温范围在其凝固点和沸点之间. 1848年,Kelvin依据卡诺定律提出了开氏温度作为物理学温标,它不依赖于任何测温物质的具体测温属性,故又称为绝对温标. 相应的温度也被称为热力学温度,以T表示,单位为开尔文,记为K.
1967年,第13届国际标度会议确立热力学温度为基本温标,并将水的三相点的热力学温度设为273.15 K. 摄氏温度与热力学温度之间的关系为
|
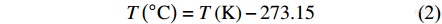
即,摄氏温度的0 °C对应热力学温度的273.15 K.
热量是物质状态发生转变的一种反映,它与人类的日常生活息息相关,很早以前人们就开始了对热的探索. 早在公元前5世纪,Empedocles[1]就提出这个世界是由气、水、土和火(热)四大元素所组成的. 一直到18世纪中叶以前,热质说(theory of caloric)盛行. 18世纪后期,人们开始通过实验证明热是粒子内部的运动. 19世纪后半期,Joule和Boltzmann等建立了统计热力学的基本原理,从而彻底推翻了传统的热质说.
由热力学第一定律可知,热是能量的一种形式,记为Q,它可以和其他形式的能量互相转化,且总能量保持不变,即:
|
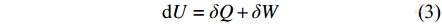
物体吸收或放出热量的能力由热容C (J·K−1)来表征,表示物体温度升高1 K所吸收的热量(单位J),
|
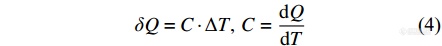
而单位质量(克,g)物体升高1 K所吸收的热量为比热容cm (J·K−1·g−1),
|
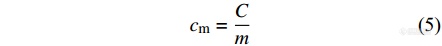
将能量表示为体积和温度的函数,则根据体积不变的条件可以得到
|
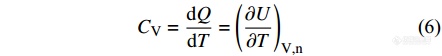
同样可以将能量表示为压强、温度的函数, 在压强不变的条件下,可得到
|
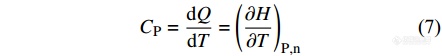
其中,H为定义的一个态函数,称为焓(enthalpy). 它与内能的关系为
|
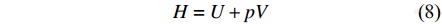
由此得到等容热容和等压热容的关系为
|
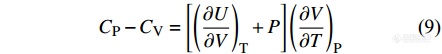
1.2 热分析(thermal analysis)
广义上来说,所有控制温度的测量过程都可以称为热分析. 1999年,国际热分析和量热协会(International Confederation for Thermal Analysis and Calorimetry, ICTAC)和美国材料与试验协会(American Society for Testing and Materials, ASTM)[2~4]对热分析的定义为:在程序温度下,测量物质的物理性质与温度或时间关系的一类技术. (A group of techniques in which a physical property of a substance is measured as a function of temperature or time while the substance is subjected to a controlled-temperature program.)
常见的热分析所测量的物理性质包括质量、温差、热量、应力和应变等. 按照测量性质的不同,最基本的热分析包括以下几种:差热分析法(differential thermal analysis, DTA)、示差扫描量热法(differential scanning calorimetry, DSC)、热机械法(thermomechanical analysis, TMA)、热重分析法(thermogravimetric analysis, TGA)等等.
示差扫描量热法(DSC)的定义是:在程序控温和稳态保护气氛下,测量进出样品和参比物之间的热流差随温度或时间变化的一种技术. 它是目前应用最为广泛的一种热分析技术. 随着科学技术的进步,DSC也得到了不断的发展,特别是近年来取得了显著的进展. 其中一个主要的进展是在20世纪90年代出现的温度调制DSC (temperature-modulated DSC, TMDSC). TMDSC在传统DSC线性扫描速率的基础上加入了调制升降温速率,可测得非线性调制热流信号,对该热流信号进行解调制,可以将总热流信号区分为可逆信号和不可逆信号两部分. TMDSC还可以通过对等温过程施加微量调制升降温速率进行准等温实验,追踪实验过程中的不可逆过程随时间的演化,并最终获得平衡状态下的可逆热容. DSC技术的另一个重要进展是近年来发展起来的闪速示差扫描量热法(fast-scan chip-calorimetry, FSC). FSC其商业化版本为Flash DSC,是基于芯片量热技术和微制造技术而发明的超快速示差扫描量热技术,它可达到106 K·s−1的扫描速率,具有较高的灵敏度,进一步将DSC的表征时间和温度窗口拓展到了发生较快速热转变的区间,增强了其表征和研究各种热转变动力学的能力.
2. 示差扫描量热法
2.1 基本原理
示差扫描量热法起源于19世纪中期. 1887年,Le Chatelier[5,6]采用热电偶首次记录了陶土的温度随时间变化的升温曲线. 1899年Roberts-Austen[7]使用参比热电偶,首次测量了样品与参比物之间的温差,发展了差热分析法(DTA). 然而这种方法只能用于定性测量样品和参比物之间的温差ΔT.
1955年,Boersma[8] 改进了DTA设备并建立了一个定量DTA测量单元,该仪器的热阻与试样无关. 对仪器的热容进行校正,可使得扫描过程中样品的热流与温差呈稳定的线性关系,从而可以定量测量热流. 这一发现最终导致了热流型DSC的诞生. 热流型DSC保留了差热分析法引入的参比物,并监测试样和参比物之间的热流差变化,得到了比只测定试样的绝对热流变化更为精确的测试结果,这也是示差扫描量热法中“示差”的含义及来源. 1964年,Watson等[9,10]提出了功率补偿型DSC的概念,这一概念有利于提高DSC的升降温速率. 此后,DSC技术不断发展并成为热分析领域的常规分析手段. 目前,市场化的DSC设备根据加热方法和测量原理主要分为热流型示差扫描量热仪(heat flux DSC)和功率补偿型示差扫描量热仪(power compensation DSC)两类[11].
热流型DSC的测试装置如图1所示.
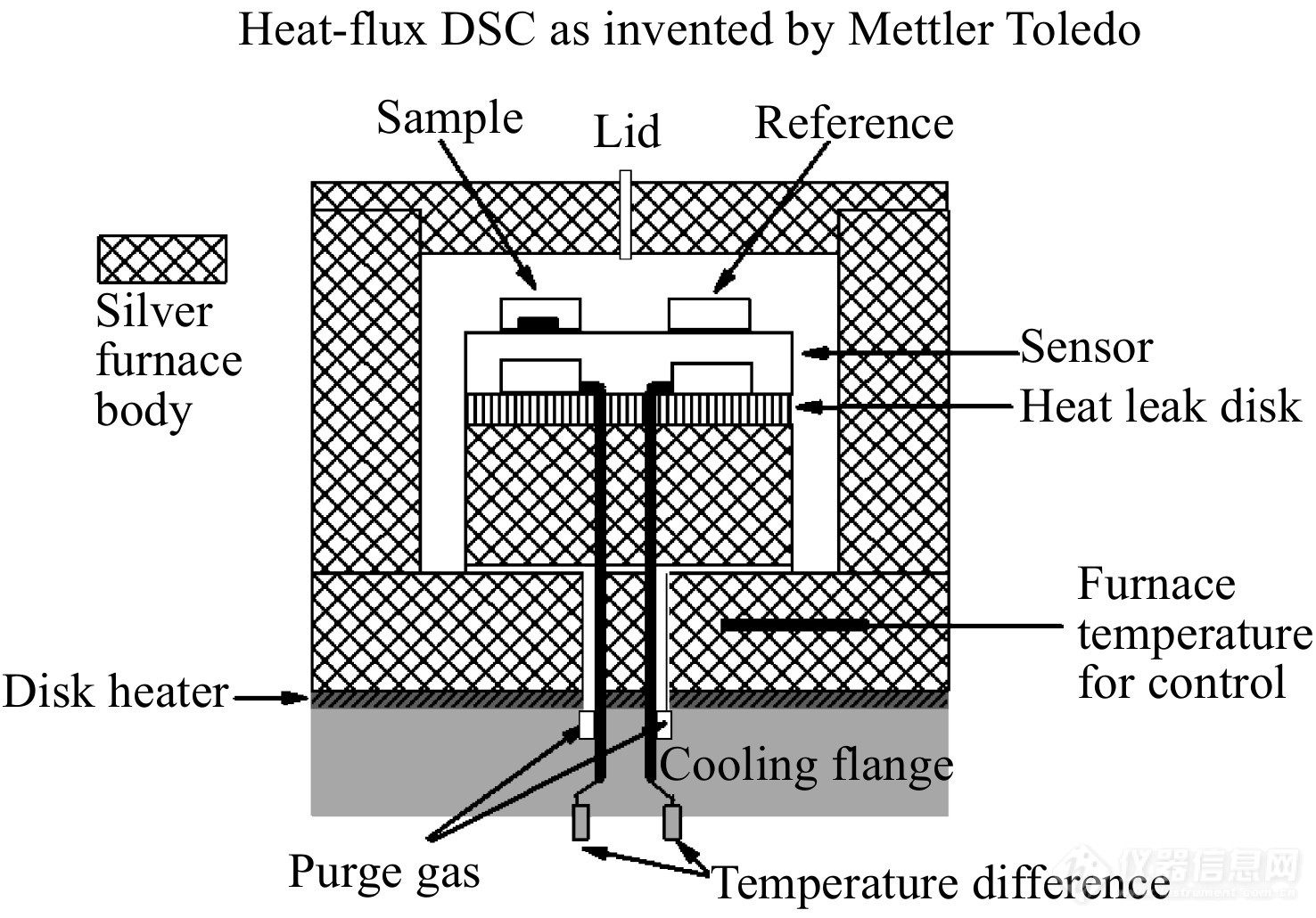
图 1
Figure 1. Illustration of heat-flux DSC (Mettler-Toledo heat-flux DSC) with the heating rate controlled through the furnace temperature. There are two sets of thermocouples measuring the heat flow between the furnace and the pan for sample and reference and two central terminals bringing the average T signal from all the thermocouples out to the computer.
热流型DSC从外部加热整个炉体,并给样品和参比物提供同样的加热功率. 由热欧姆定律可知,由炉体流到试样坩埚的热流[Math Processing Error]ϕs 以及由炉体流入参比坩埚的热流[Math Processing Error]ϕr分别为[12]
|
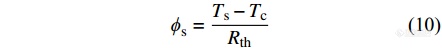
|
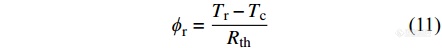
其中,[Math Processing Error]Ts、[Math Processing Error]Tr和[Math Processing Error]Tc分别为试样温度、参比温度和炉体温度,[Math Processing Error]Rth为热阻.
DSC检测信号[Math Processing Error]ϕ为2个热流之差,
|
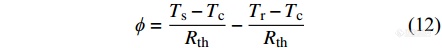
由于参比坩埚和试样坩埚相同,仪器两边具有对称性,可将上式简化为
|
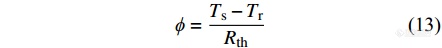
即,热流型DSC的检测信号[Math Processing Error]ϕ与试样和参比物之间的温差[Math Processing Error]ΔT=Ts−Tr成正比.
热流型DSC对整个炉体进行加热,测试氛围均匀且稳定,因此能保持较为稳定的基线. 另一方面,炉体的热容较大,不利于快速升降温,因此热流型DSC的升降温速率较慢.
功率补偿型DSC的测试装置如图2所示.
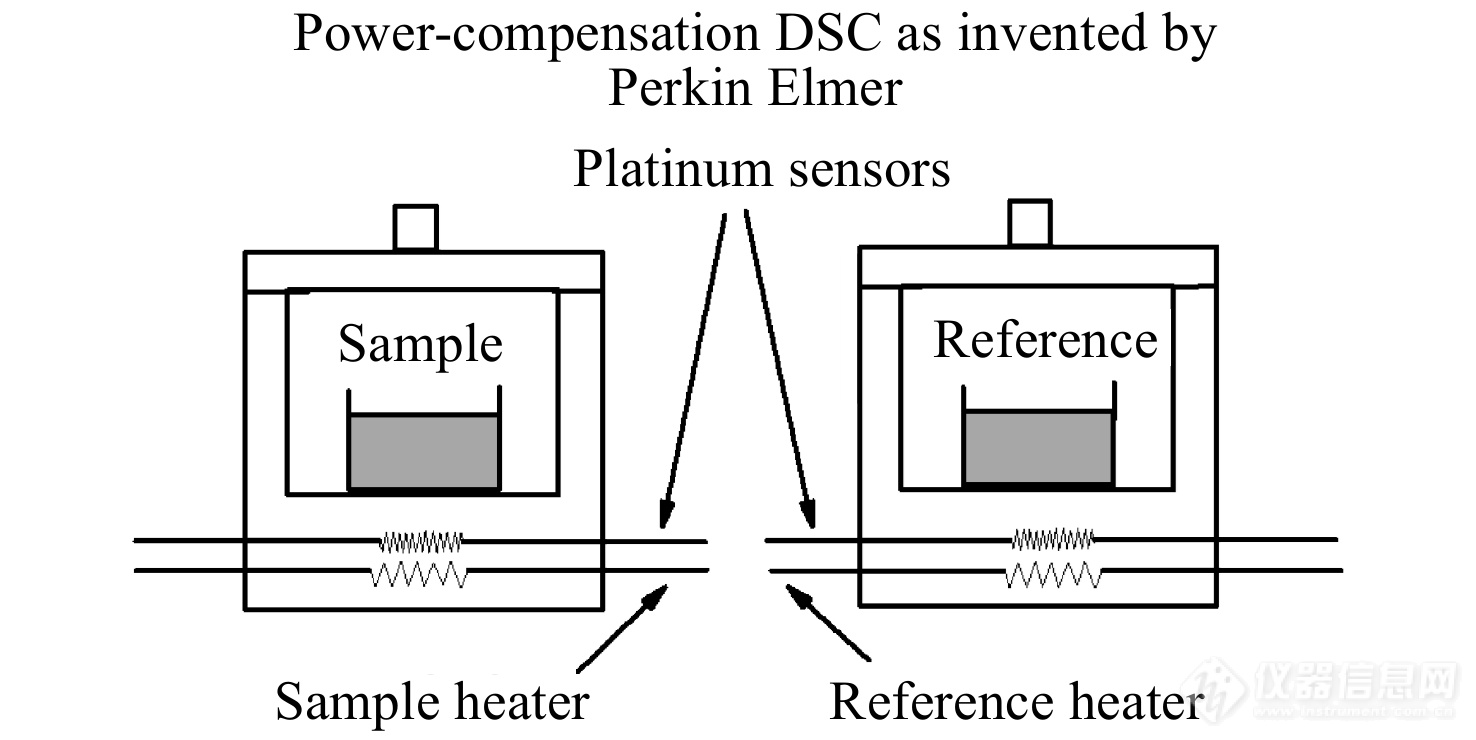
图 2
Figure 2. Illustration of power-compensation DSC as invented by Perkin Elmer with the reference and the sample separately heated by two platinum resistance thermometers in two calorimeters mounted in a constant temperature block.
功率补偿型DSC采用2个独立的加热器分别对样品盘和参比盘进行控温和功率补偿,当样品发生吸热或者放热效应而导致样品与参比物之间的温差不为零时,电热丝将及时对参比盘或样品盘输入电功率以进行热量补偿,使两者的温度始终处于动态零位平衡状态,同时记录样品和参比物的2只补偿电热丝的功率之差随时间的变化关系,
|
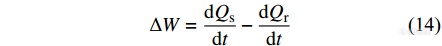
功率补偿型DSC的热源更贴近样品,温度响应灵敏,因此升降温速率更快. 为了准确测量样品的热效应,功率补偿型DSC的2个炉体必须具有很高的对称性,然而仪器内部的环境往往会随着时间而发生改变,因此功率补偿型DSC的基线容易发生漂移,不如热流型DSC稳定.
2.2 实验技巧
2.2.1 仪器校准
首先采用标准物质在待测温度范围内对仪器进行校准,以保证测量值与参考值相吻合. 校准的内容主要包括DSC曲线上的温度值以及热流速率值. 因此标准物质应具有较好的稳定性,其测量性能必须具有可靠的文献参考值. 常用于校准的标准物质有铟、锡、尿素、苯甲酸等等,这些标准物质可用于不同温度范围内的校准. 图3是采用铟进行熔点以及熔融焓校准得到的测量结果,将标准物质的熔点以及熔融焓的测量值与文献参考值进行比较,若测量值不在误差限之内,则需要对仪器的参数进行调整,使测量值与参考值相符合[13].
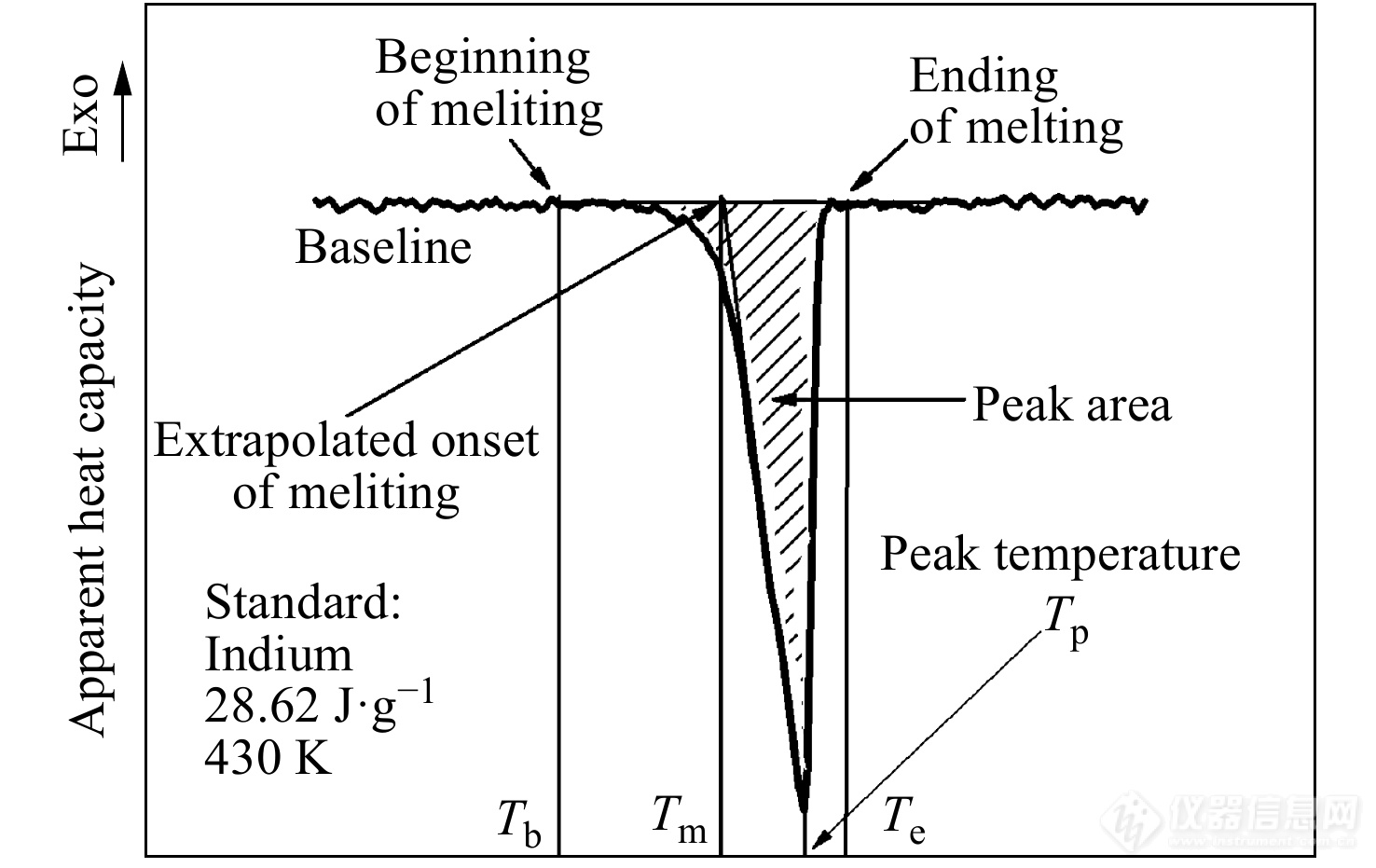
图 3
Figure 3. Illustration of the calibration of temperature and heat-flow rate with the standard material Indium for DSC measurement. The curve is characterized by its baseline and the endothermic process with some characteristic temperatures including the beginning of melting, Tb, the extrapolated onset of melting, Tm, the peak temperature, Tp, and the end of melting where the baseline is finally recovered, Te. Generally, Tm is the most reproducible point as an accurate measure of the equilibrium temperature which are used for the temperature calibration. The peak area below the baseline can be compared with the expected fusion heat of standard materials for the calibration of the heat flow rate.
2.2.2 样品制备
DSC实验采用坩埚作为试样容器,包括铝坩锅、高压坩埚以及具有特殊用途但使用较少的铂金、黄金、铜、蓝宝石或者玻璃坩埚等等. 其中最常用的是铝坩埚,包括40 μL标准铝坩埚和20 μL轻质铝坩埚. 带盖的40 μL标准铝坩埚应用范围较广,能进行固体和液体样品的测试. 20 μL的轻质铝坩埚的热容较小,有利于提高测试信号的分辨率和灵敏度,可用于质量较小的薄膜或者粉末样品的测试,一般不用于液体样品的测试. 称量样品之前首先需要选取2个质量十分相近的坩埚,以保证DSC仪器具有较好的对称性. 此外,取放坩埚时采用镊子夹取坩埚,并将坩埚放置在称量纸上,以免污染坩埚及坩埚内的样品.
然后选择样品质量. 一般来说,样品质量越少越好,较少的样品量可以减小样品内部的温度梯度,提高信号的分辨率,此外还能保证与坩埚底部的良好接触,有利于提高基线的稳定性和温度测量的准确度. 然而样品质量过少会导致信号的灵敏度较低. 因此,在称量样品时需要综合考虑两者的影响. 通常,样品的体积不超过坩埚体积的2/3,有机样品的质量为5~10 mg,无机样品的质量为10~50 mg[12]. 称量时采用差减法,先用分析天平称量空坩埚的质量,然后放入样品,称量样品和坩埚的质量之和,两者相减则得到样品的质量. 称量时每个质量都需要测量3遍,保证质量称量的准确度在±0.2%.
装样过程需要注意3个方面. 一是保证样品与坩埚之间具有良好的热接触,以提高信号的分辨率和测试结果的可重复性. 这要求样品具有较平的底部,最好是细粉末状或者是平整的薄片. 若样品底部不规则,可以用20 μL的轻质铝坩埚的坩埚盖将样品压在坩埚底部,或者将样品研磨成粉末. 二是注意不要污染坩埚. 残留在坩埚表面的样品很有可能会污染传感器,导致一些信号假象,并且会使热传导变差. 三是选样应具有代表性并保证样品的均匀性.
装样完成之后盖上坩埚盖,并在盖子上钻一个大孔(>1 mm),或者多钻几个小孔. 这样做的目的,一是形成一个自由扩散的气氛,二是防止样品在加热过程中因体积膨胀而掀翻盖子溅出坩埚,污染传感器[12].
2.2.3 温度程序
在设计温度程序时需要选择合适的温度范围和升降温速率. 在终点温度不超过样品的分解温度的前提下,扫描的温度范围应该足够宽,以保证能检测出所有目标热效应的热流信号,同时保证在热效应之前和之后的热流曲线具有较平稳的基线. 升降温速率的快慢会影响测试曲线的峰形和转变温度等. 较快的升温速率有利于提高测试灵敏度和效率,但会导致峰形变宽. 而较慢的升温速率可提高测试的分辨率. 传统DSC的升降温速率范围通常在0.1~250 K·min−1之内,使用不同的制冷机可得到不同的扫描速率范围,常用的升降温速率在10~20 K·min−1左右. 设计温度程序时还需要在升降温片段的两端加上时间较短(2 s)的等温片段,以保证样品在升降温扫描之前已经达到了稳态. 通常需要将设计的温度程序重复试验几次,确保测试结果的可重复性[13].
2.2.4 保护气氛
DSC测量需要往炉体内通入某一恒定流速的气体以形成特定的稳态气氛. 气氛可以为惰性的、反应性的或者腐蚀性的,在不同的气氛条件下测量可获得不同的测试信息. 通入惰性气体可以防止测试过程中发生水气凝结,污染物沉积,高温氧化等现象. 常用的高纯度惰性气体有氮气、氦气和氩气等. 氮气是最常用的保护气氛,它在约600 °C以下都是惰性的,并且具有较好的热传导能力,能得到分辨率和灵敏度较好的实验结果. 氩气常用于金属的高温测试. 氦气的热传导性能最好,在DSC测试中常被用于提高信号时间常数以及低温区的测量. 测试过程中调节减压阀,保证气体流速平稳,使实验结果具有较好的重现性. 通常气体的流速为20~100 mL·min−1,最常用的为50 mL·min−1[14]. 当需要通入反应性或者腐蚀性气体时,应注意操作的规范性,减小气体对仪器的腐蚀和伤害,保证所有的安全措施都到位.
在使用仪器的过程中需要开启制冷机,保证有稳定的冷源作为参考温度源,以提高信号曲线的可重复性. 制冷机使用结束之后,需要进行除水操作,以免水分残留在仪器内,造成测试结果不稳定.
2.3 应用举例
2.3.1 比热容
DSC一般采用三段法测量样品的比热容[15]. 以相同的扫描速率进行如下3次实验:(1) 样品盘和参比盘上分别摆放一个空坩埚,进行空白实验,得到空白信号[Math Processing Error]φempty(T). (2) 将标准物质蓝宝石放入试样盘的空坩埚中,参比盘保持原先空坩埚,测量得到参比信号[Math Processing Error]φsapphire(T). (3) 将样品放入试样盘的空坩埚中,参比盘保持原先空坩埚,测量得到样品信号[Math Processing Error]φsample(T).
图4是采用三段法测量比热容的热流曲线示意图.
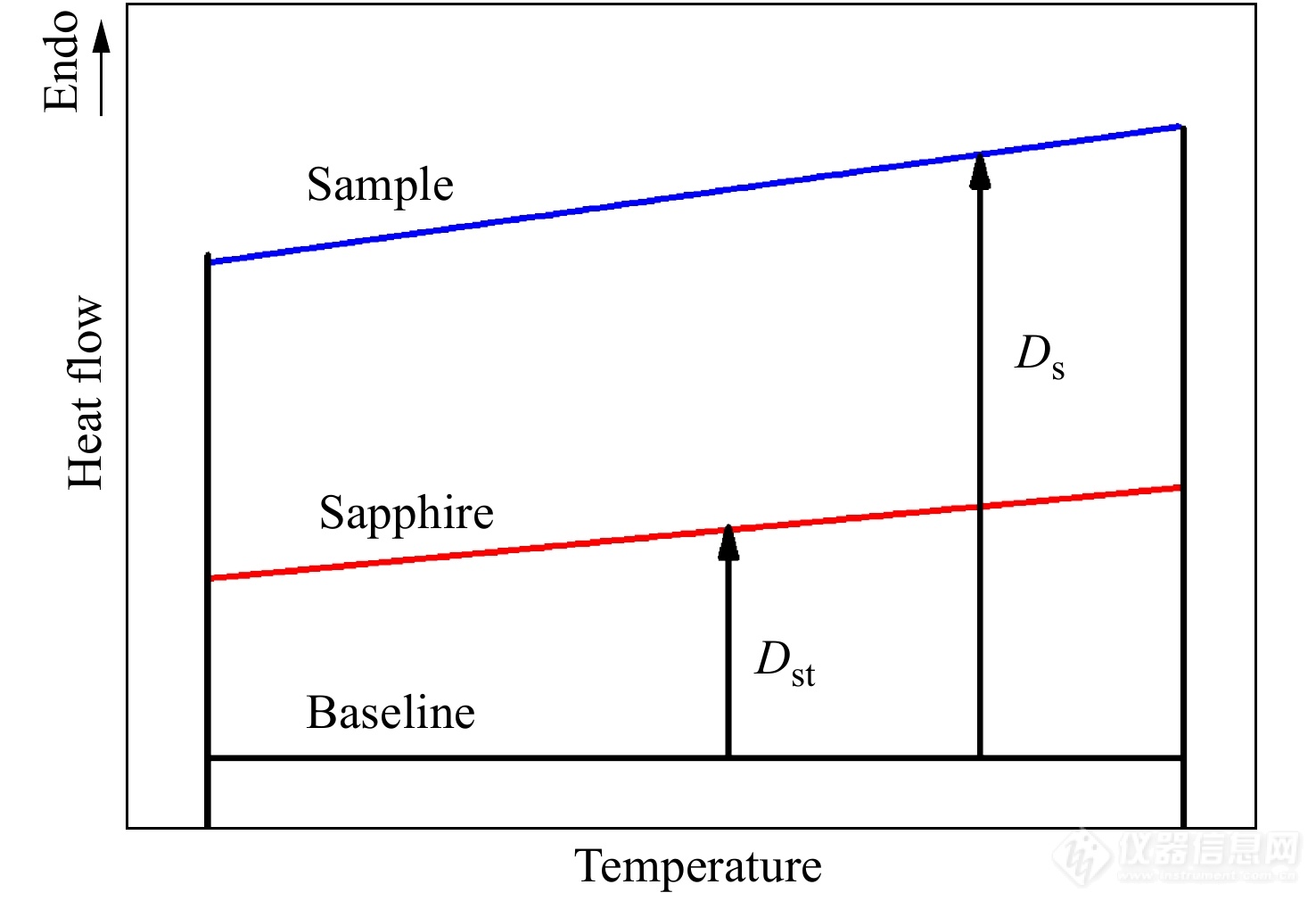
图 4
Figure 4. Heat flow curves of standard sapphire and unknown specimens where Ds (mW) is the vertical displacement between the baseline and the specimen DSC thermal curves at a given temperature while Dst (mW) is vertical displacement between the baseline and the sapphire DSC thermal curves at a given temperature.
由蓝宝石的比热容[Math Processing Error]cm,sapphire、样品和蓝宝石的质量[Math Processing Error]m可求出样品的比热容:
|
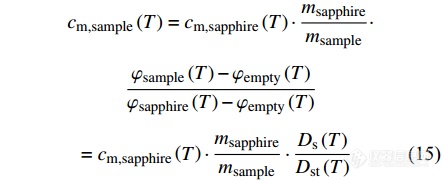
更多的有关高分子标准热容数据可从ATHAS (Advanced THermal AnalysiS)[16]等数据库中查找.
2.3.2 热转变温度
高分子材料的物理热转变温度主要包括玻璃化温度和熔点. 玻璃化温度[Math Processing Error]Tg是非晶态聚合物在玻璃态和高弹态之间转变的温度. 研究玻璃化转变温度可以得到有关样品的热历史、稳定性、化学反应程度等重要信息,对于实验研究、质量检测等具有重要意义. 玻璃化转变温度通常取DSC曲线发生玻璃化转变台阶上下范围的中点. 图5是ASTM方法[17]测量聚合物玻璃化转变温度的热流曲线图,在台阶的拐点[Math Processing Error]Ti处做一条切线,由这条切线与基线的交点可得到外推起始温度[Math Processing Error]Tb1和外推终止温度[Math Processing Error]Te1,这两点的中点即为玻璃化转变温度[Math Processing Error]Tg.
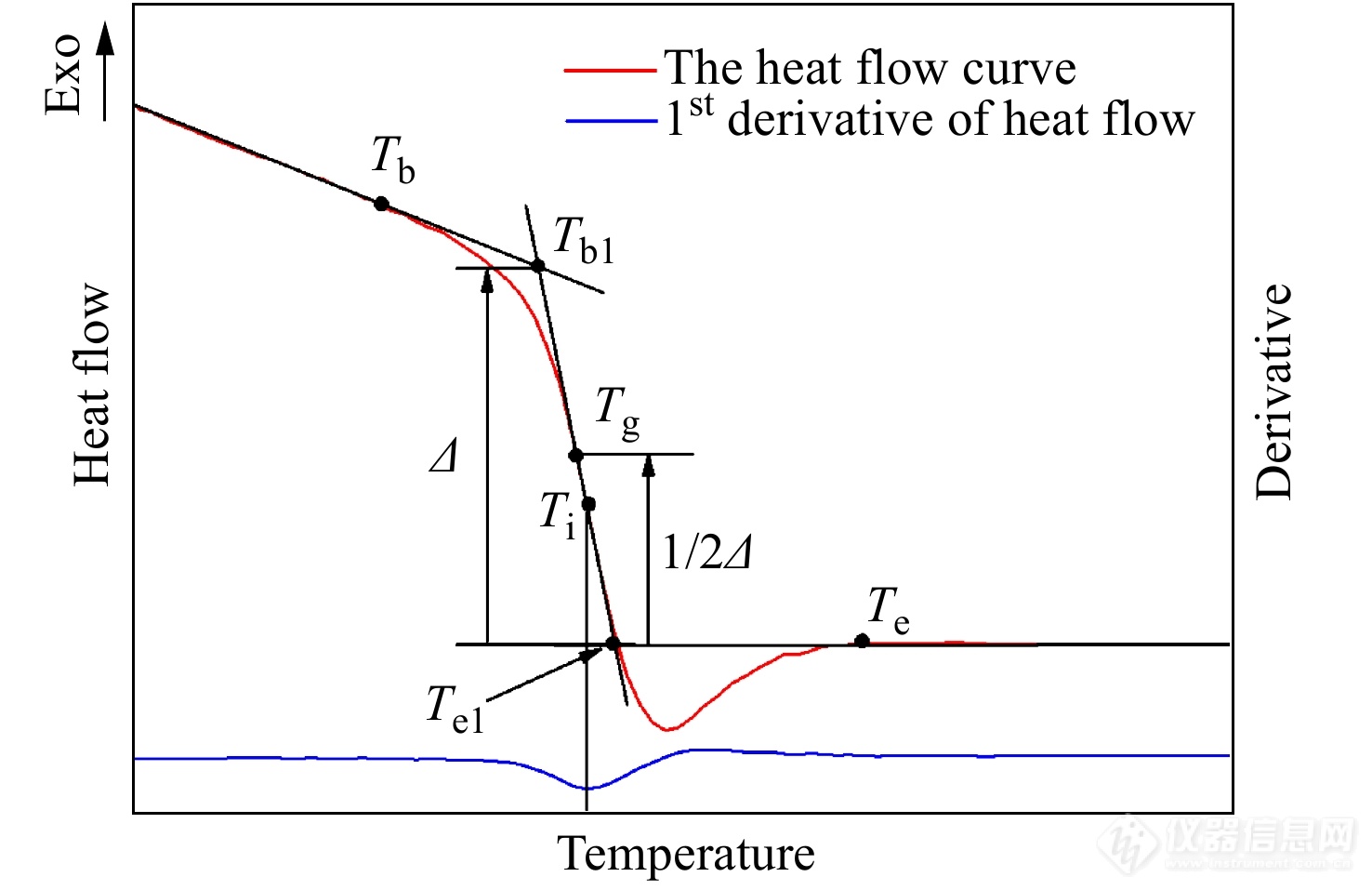
图 5
Figure 5. The heat-flow rate (the upper curve in the left axis) and its derivative (the lower curve in the right axis) curves in the glass transition region with some characteristic temperatures including the beginning of glass transition Tb, the extrapolated onset temperature Tb1, the midpoint temperature Tg, the inflection temperature Ti, the extrapolated end temperature Te1 and the temperature of return-to-baseline Te as listed. The glass transition is determined by Tg (°C)—the point on the thermal curve corresponding to the half of the heat flow difference between the extrapolated onset and extrapolated end.
玻璃化转变温度与升降温速率、杂质、样品尺寸等有关. 因此,测试结果应该标注测量时的升降温速率.
小分子一般取熔融峰前端的延长线与基线的交点,即熔融起点作为熔点. 然而高分子化合物具有较宽的片晶厚度分布,因而具有较宽的熔程,导致其熔点的测量方法与小分子化合物不同. 一般取高分子熔融峰的峰顶点温度作为熔点.
2.3.3 转变焓
DSC的一个重要用途就是测量聚合物的转变焓,包括熔融焓、结晶焓、反应焓等等.
转变焓一般是通过对DSC热流曲线峰面积进行积分得到的.
|
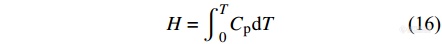
当转变峰曲线左右两边的基线水平时,可通过直接连接转变前后的基线进行面积积分. 当聚合物的熔程较宽或者基线发生较大偏移时,简单的基线法无法较为准确地计算转变焓. 此时,可根据相转变过程中吸收的熔融热的多少来确定基线的位置,也可简单地根据峰顶的位置将熔融峰分成左右两部分,两边使用各自的基线来加和计算[11]. 更多的定量计算可通过计算机程序[18]或者去卷积[19]计算得到.
2.3.4 DSC与其他技术连用
随着红外光谱仪(infrared spectrometer, IR)、X射线衍射(X-ray diffraction, XRD)、色谱等常规技术的不断发展,DSC技术与常规技术的连用成为了目前高分子研究的方向之一. 通过结合多种表征技术的优势,可以获得高分子样品在相转变以及反应过程中的形貌结构、组成成分、热性能、机械性能等多种信息,帮助研究者从多个角度、更深层次地理解高分子在热转变过程中的内在机理.
DSC与X射线衍射、原子力显微镜(atomic force microscopy, AFM)、拉曼光谱等技术的连用被广泛应用于研究高分子的相转变机理,包括晶体结构的相转变[20]、嵌段共聚物中的微相分离与结晶的相互作用[21],以及共混物中的分级结晶行为[22]等. 高分子在实际加工过程中不仅要进行退火等热处理,通常还会在拉伸场和剪切场下进行取向. 因此,将DSC与动态热机械分析(dynamic mechanical analysis, DMA)等技术连用有助于推进对高分子的聚集态结构在拉伸和取向状态下随温度变化的相关研究[23].
3. 温度调制示差扫描量热法
3.1 基本原理
DSC样品的热流信号[Math Processing Error]ϕ可分为显热流[Math Processing Error]ϕsens和潜热流[Math Processing Error]ϕlat 2个部分[24].
|
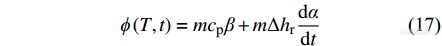
其中,[Math Processing Error]mcpβ为显热流,对应于样品的比热容,它依赖于样品的升降温速率. [Math Processing Error]mΔhrdαdt为潜热流,对应于样品中的物理化学过程,如化学反应、结晶过程或蒸发过程等等,它依赖于远离平衡态的内部变量的变化,不具有很强的升降温速率依赖性. 然而,潜热流所带来的样品组分变化会影响显热流所对应的比热容,传统DSC只能测定总热流随温度或时间的变化,无法有效地区分潜热流和显热流. 另外,传统DSC也无法测量等温过程的比热容.
为了解决上述问题,人们注意到显热流给出的是一个可逆信号,而潜热流大多反映不可逆热过程,于是在线性温度程序上叠加一个很小的调制温度,来区分可逆和不可逆热流信号,由此发明了温度调制示差扫描量热法(TMDSC). 早在20世纪初,温度调制技术就被应用到了量热研究中. 1910年,Corbino[25,26]发展了调制量热仪的理论,并首次采用3ω法(third-harmonic method)[27]测量了导电铁丝的热容. 20世纪60年代,由于实验技术的进步,调制量热法取得了相当大的进展,Kraftmakher[28]、Sullivan和Seidel[29]开始提出AC量热法. 1971年,Gobrecht[30]等采用DSC直接测量出无机聚合物在玻璃化转变处的频率依赖的复合热容,这可以被认为是首次TMDSC实验. 直到1992年,Reading在第九届北美热分析会上正式提出温度调制示差扫描量热法[31~34],随后美国TA公司推出首个调制DSC的专利技术,称为MDSC. 此后,随着计算机技术的进步,各家热分析供应商相继推出类似的温度调制程序专利技术,TMDSC成为热分析领域的标准工具并被广泛应用于聚合物分析表征研究.
通过引入一个调制温度,TMDSC在较慢的线性升温速率的基础之上获得了一个瞬间的剧烈温度变化,从而得到兼具较高的灵敏度和分辨率的热流信号,能实现重叠热效应的有效分离以及准等温过程可逆热容的测量.
目前最常用的TMDSC是正弦波模式温度调制,其温度程序为,
|
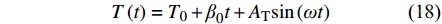
其中,[Math Processing Error]T0为开始温度,[Math Processing Error]β0为基础升温速率,AT为温度振幅,[Math Processing Error]ω=2πtp为调制频率,[Math Processing Error]tp为调制温度周期. 图6和图7分别展示了正弦波形TMDSC的温度程序以及实验测得的热流信号,该调制热流信号是对温度程序的正弦同步响应,其相对温度程序有相位差[Math Processing Error]φ的滞后.
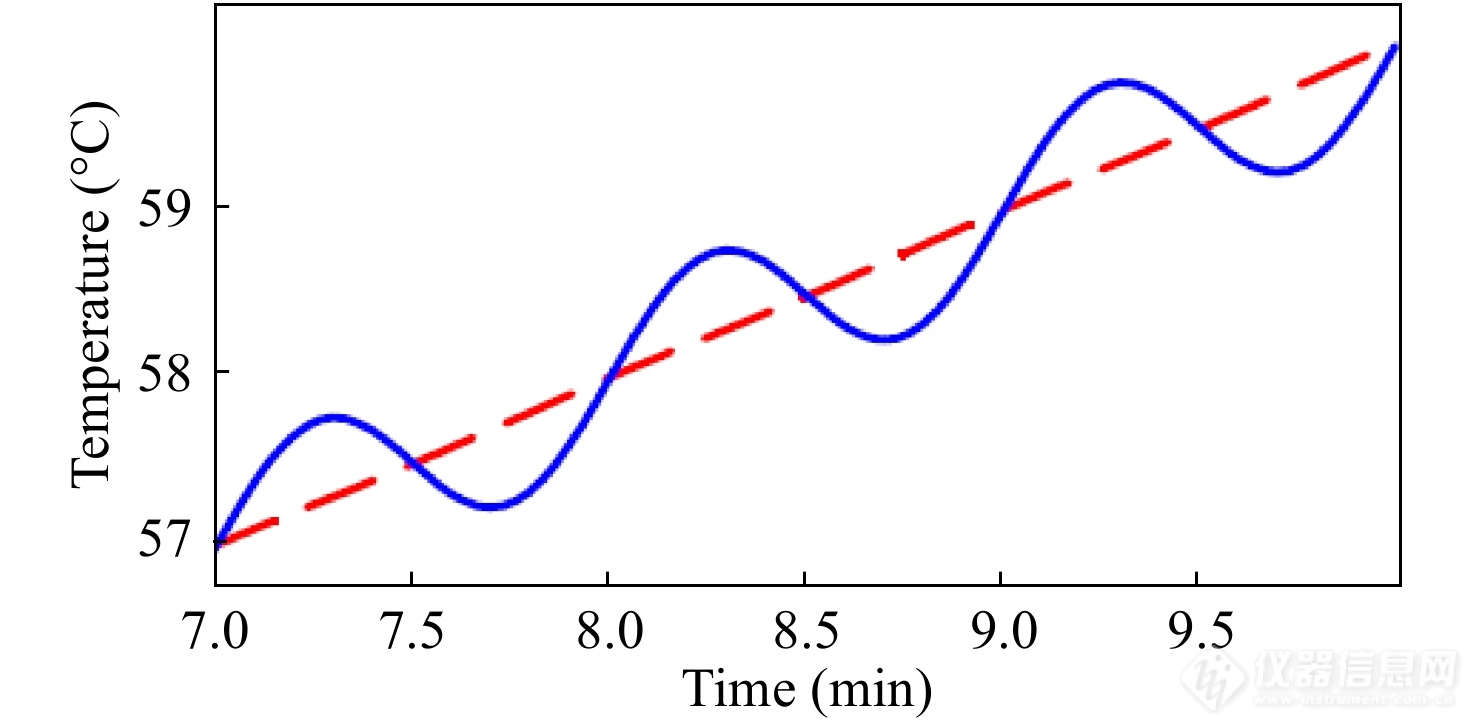
图 6
Figure 6. Typical temperature profile of sinusoidal TMDSC (blue and solid curve) and its underlying heating rate curve with [Math Processing Error]β0 of 1 K·min−1 (red and dashed line). The amplitude of modulation AT is 0.5 K, the period of modulation [Math Processing Error]tp is 60 s. (Reprinted with permission from Ref.[24]; Copyright (2009) Polymer Bulletin)
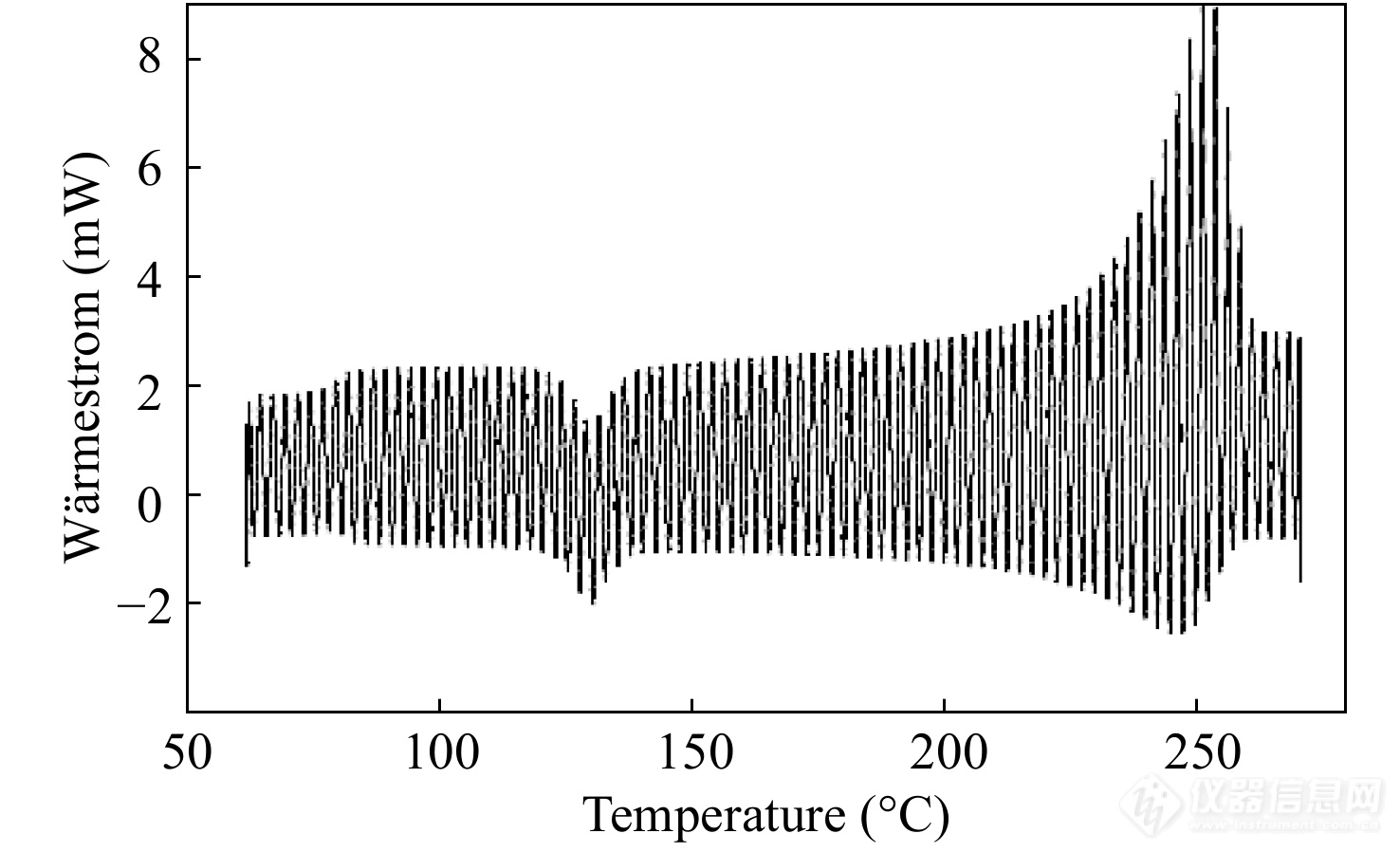
图 7
Figure 7. The heat-flow curve measured by the sinusoidal temperature-modulated DSC (Reprinted with permission from Ref.[24]; Copyright (2009) Polymer Bulletin)
对图7中热响应信号进行平均化计算得到总热流[Math Processing Error]⟨ϕ(t)⟩曲线如图8所示. 总热流曲线相当于常规DSC曲线,由总热流可求出总热容.
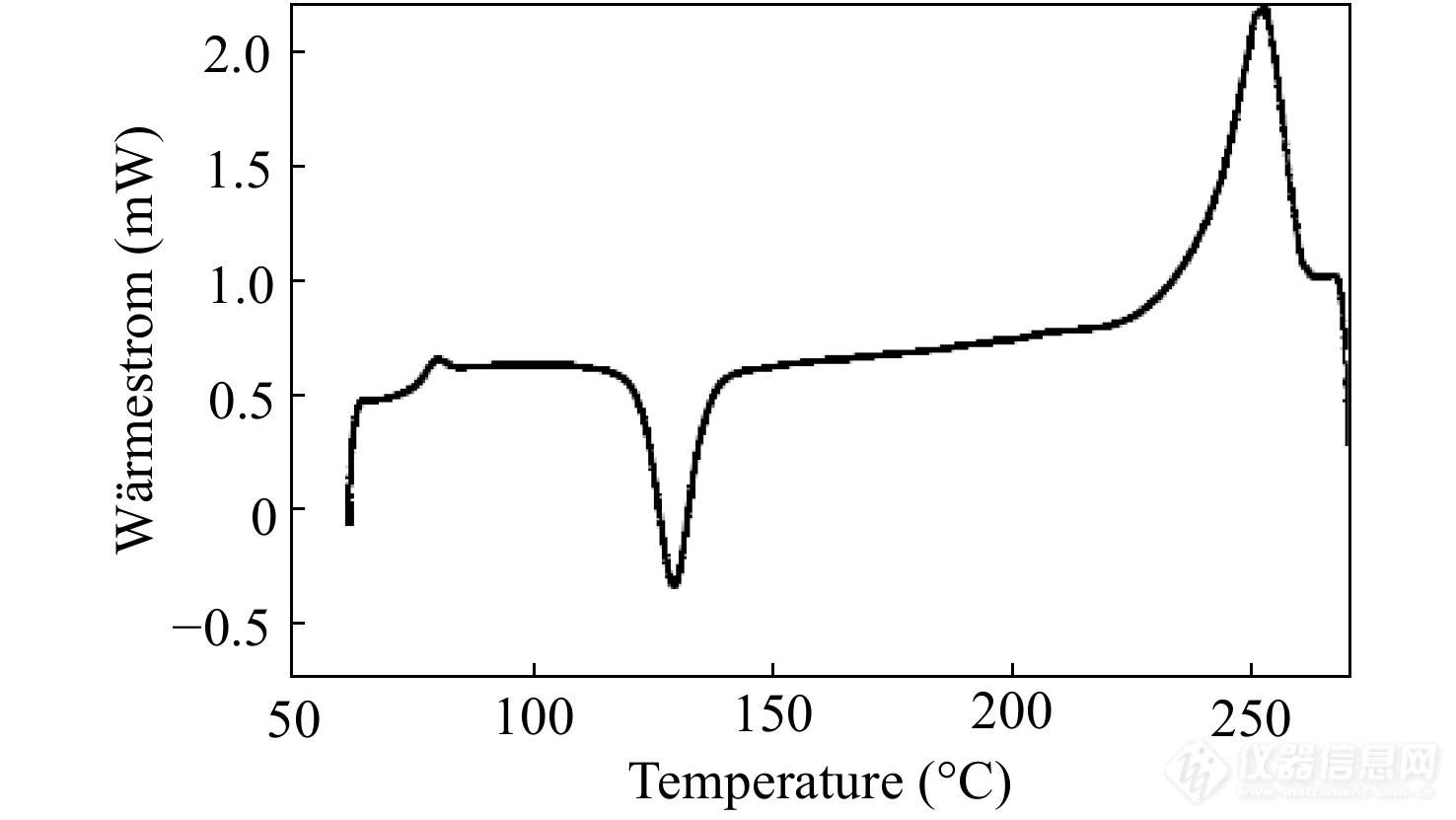
图 8
Figure 8. The total heat-flow curve of a sinusoidal TMDSC curve. (Reprinted with permission from Ref.[24]; Copyright (2009) Polymer Bulletin)
|
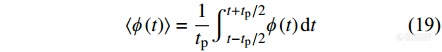
进一步采用离散傅里叶变换对图7曲线去卷积分析,
|
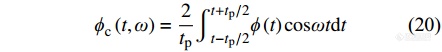
|
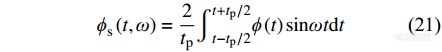
其中,[Math Processing Error]ϕc(t)是对加热速率无滞后的周期性热流分量,由[Math Processing Error]ϕc(t,ω)可计算可逆热流:
|
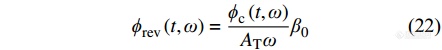
由总热流减去可逆热流即可得到不可逆热流:
|
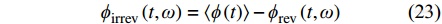
另一种常见的TMDSC为锯齿形TMDSC,其温度程序为:
|
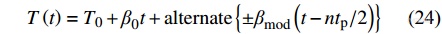
图9展示了锯齿形DSC的温度程序图,其中,[Math Processing Error]T0=0∘C,[Math Processing Error]β0=1 K·min−1,[Math Processing Error]βmod=3 K·min−1,[Math Processing Error]tp=60s.
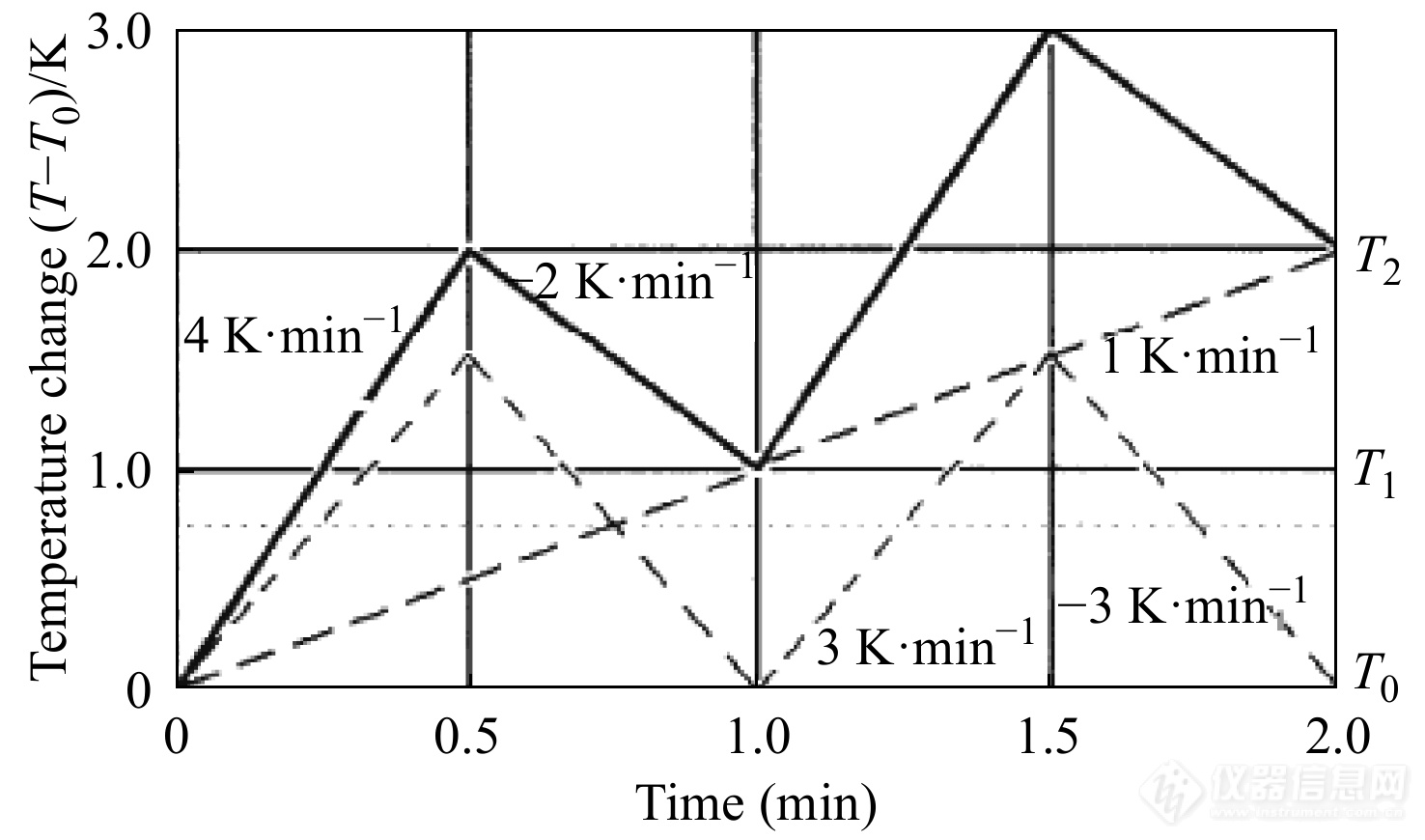
图 9
Figure 9. Typical temperature profile for sawtooth TMDSC (solid line) and its deconvoluted underlying heating rate [Math Processing Error]β0 of 1 K·min−1 and the reversing rate of temperature change of ±3 K·min−1 (dashed lines). T1 and T2 indicate the beginnings and ends of the cycles, respectively. (Reprinted with permission from Ref.[35]; Copyright (2014) Springer Nature)
图10展示了由锯齿形TMDSC得到的线性即时响应的热流信号图. 一般来说锯齿形TMDSC的升降温程序比较长,可提供足够的反应时间以保证样品在由升温转换为降温(或者由降温转换为升温)之前达到稳态. 一般每个升温或降温片段都需要至少30 s的仪器调整时间来达到热响应信号的稳定值,如图11所示. 因此,在数值计算时,只需要取热流信号接近于上限或者下限的那部分数据,并将热流信号延长至升温或者降温片段的起始处,即可得到如图10所示的热流信号. 可在锯齿形TMDSC程序中间隔插入等温程序,以检测体系是否达到稳态以及基线的平稳性.
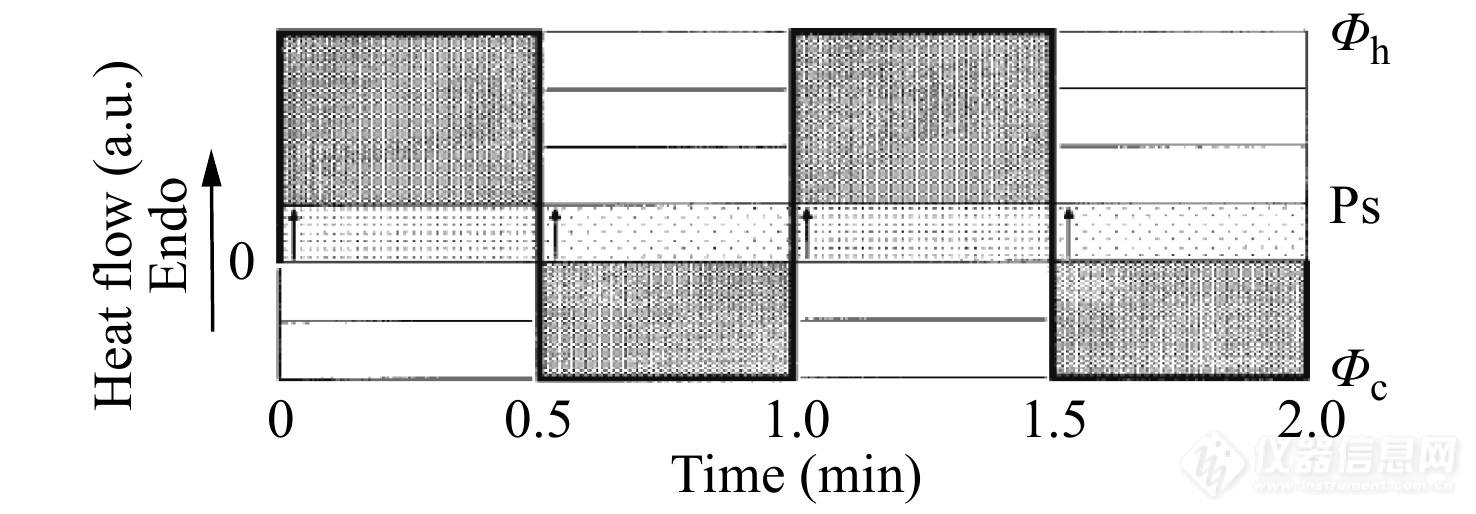
图 10
Figure 10. Illustration of the linear thermal response (solid lines) for the temperature profile of Fig 11. The lightly dotted boxes and the heavily dotted boxes separately indicate the underlying and the reversing responses. The heavy line represents the heat flow rate [Math Processing Error]ϕ(t). The pseudo-isothermal level (Ps), the zero level (0) and the value of upper and lower limits of the heat flow rate [Math Processing Error]ϕh and [Math Processing Error]ϕc are marked, respectively. (Reprinted with permission from Ref.[35]; Copyright (2014) Springer Nature)
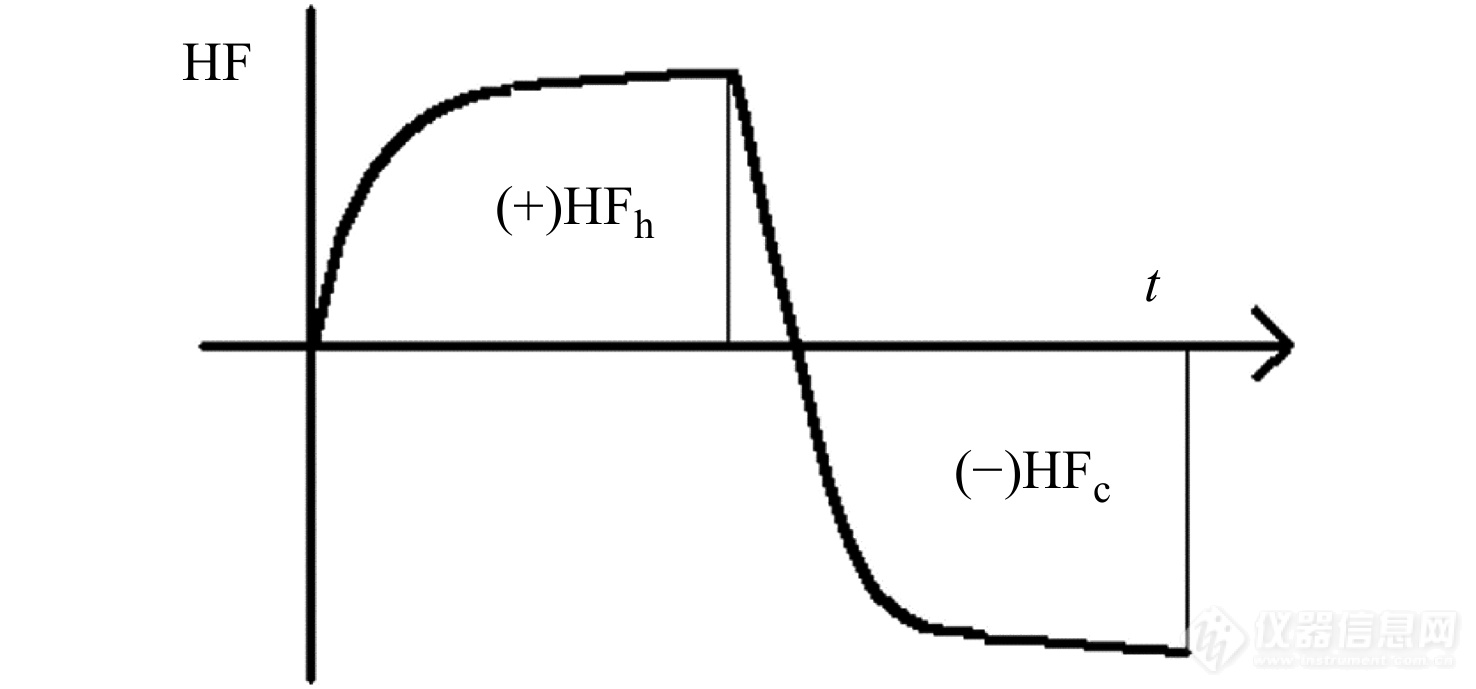
图 11
Figure 11. Illustration of the nonlinear thermal response in each cycle measured by sawtooth TMDSC where HFh and HFc separately represent the heat flow rate measured in the heating and cooling half cycles.
首先对锯齿形TMDSC的响应信号进行平均化计算得到基本信号[Math Processing Error]⟨HF(t)⟩,由基本信号[Math Processing Error]⟨HF(t)⟩可以求出总的热容信号:
|
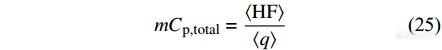
锯齿形TMDSC的热流信号无需傅里叶转变,可直接由升降温的热流信号求出可逆热容,该结果能达到与标准DSC相同的准确度.
|
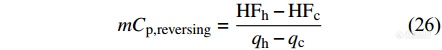
由总热容减去可逆热容求出不可逆热容
|
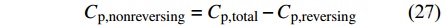
但是此处往往过高地估计了可逆热容,会导致不可逆信号成为负信号,采用常规正弦波调制时在高分子熔融峰温度范围内常常将其解读为熔融重结晶的信号,因此需要计算不平衡热容[Math Processing Error]Cp,imbalance来反映不可逆热容的真实趋势.
|
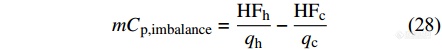
[Math Processing Error]Cp,imbalance反映了不可逆热流在升降温中的差异,对于准确解读晶体熔融等复杂过程的不可逆热容部分有重要意义[35].
3.2 实验技巧
3.2.1 样品质量
TMDSC的实验操作与常规DSC相同. TMDSC要求样品与坩埚的热传递良好,因此,样品质量和厚度越小越好. 样品质量太大会导致热滞后效应加剧,响应周期延长,测量的有效频率和振幅范围减小.
3.2.2 温度程序
正弦波模式温度调制得到的计算结果精度较高,但要求热响应信号呈线性且平稳变化,而通常实验得到的热信号会与仪器的热滞后信号耦合,影响测量的准确度. 此外,正弦波模式的傅里叶变换仅仅计算了一次谐波项,导致在有些热效应中过高地估计了可逆热流. 锯齿模式温度调制的数据处理过程更为简单可靠,测量结果可以达到普通DSC的精确度. 锯齿模式温度调制无需进行傅里叶分析,因此可以直接在时间域中分析不可逆过程以及慢热过程,保证在测试过程中样品处于稳态,避免由于基线不稳定导致的分析误差[35]. 实际测试时需要根据测试要求选择不同的温度调制模式.
TMDSC的参数有基础升温速率、调制频率以及调制振幅. TMDSC的基础升温速率较慢,通常在1~3 K·min−1,以保证热流信号具有较高的分辨率. 而调制振幅和调制频率的设置更为复杂,需要保证在测试的热效应范围内出现4~5个振荡周期. 通常温度振幅为0.5~2 K,调制周期通常为30~120 s. 调制振幅和调制频率过高时,会超出仪器的响应周期. 而当调制振幅和调制频率过低时,热流信号会受到基线漂移的影响,而且快速相转变过程中的有效调制周期数过少,信号分辨率下降.
3.3 应用举例
3.3.1 可逆热容和不可逆热容
TMDSC的一个重要应用是区分可逆热容和不可逆热容信号. 胡文兵等[35]采用锯齿形TMDSC研究PET在升温过程中发生的热效应,得到图12所示的比热容随温度变化的关系图. 其中,黑点代表的是ATHAS数据库所提供的无定形PET在不同温度下的标准比热容数据. 虚线为总比热信号,该曲线表明PET在升温过程中依次出现了玻璃化转变、冷结晶以及熔融. 而实线代表了可逆比热信号,它包括了较低温度区域的玻璃化转变和较高温度区域的熔融峰. 可逆比热曲线上的熔融峰与总比热曲线的熔融峰面积相近,说明计算得到的熔融可逆信号偏大. 由总热流信号减去可逆热流信号,得到不可逆比热信号如图中短线-点-短线符号代表的曲线所示. 除了冷结晶峰和熔融峰,不可逆比热曲线在500 K左右出现了一个向下的放热峰,这似乎表明PET在高温区发生了熔融重结晶. 进一步计算不平衡热容,得到图中细点组成的曲线. 该曲线与不可逆热容曲线相比仅出现了向上的熔融峰,说明不可逆比热曲线上高温区的负信号并非熔融重结晶. 上述结果表明,实际实验过程中的热流信号并非完全的线性和稳态,非线性热流信号与非稳态热流信号发生耦合,会导致可逆热容信号偏大,进一步将其从基线热容扣除会导致不可逆热容信号出现负值. 而锯齿形TMDSC中的不平衡热容能够避免不可逆热容负值的出现,更为正确地反映不可逆热容的偏移方向.
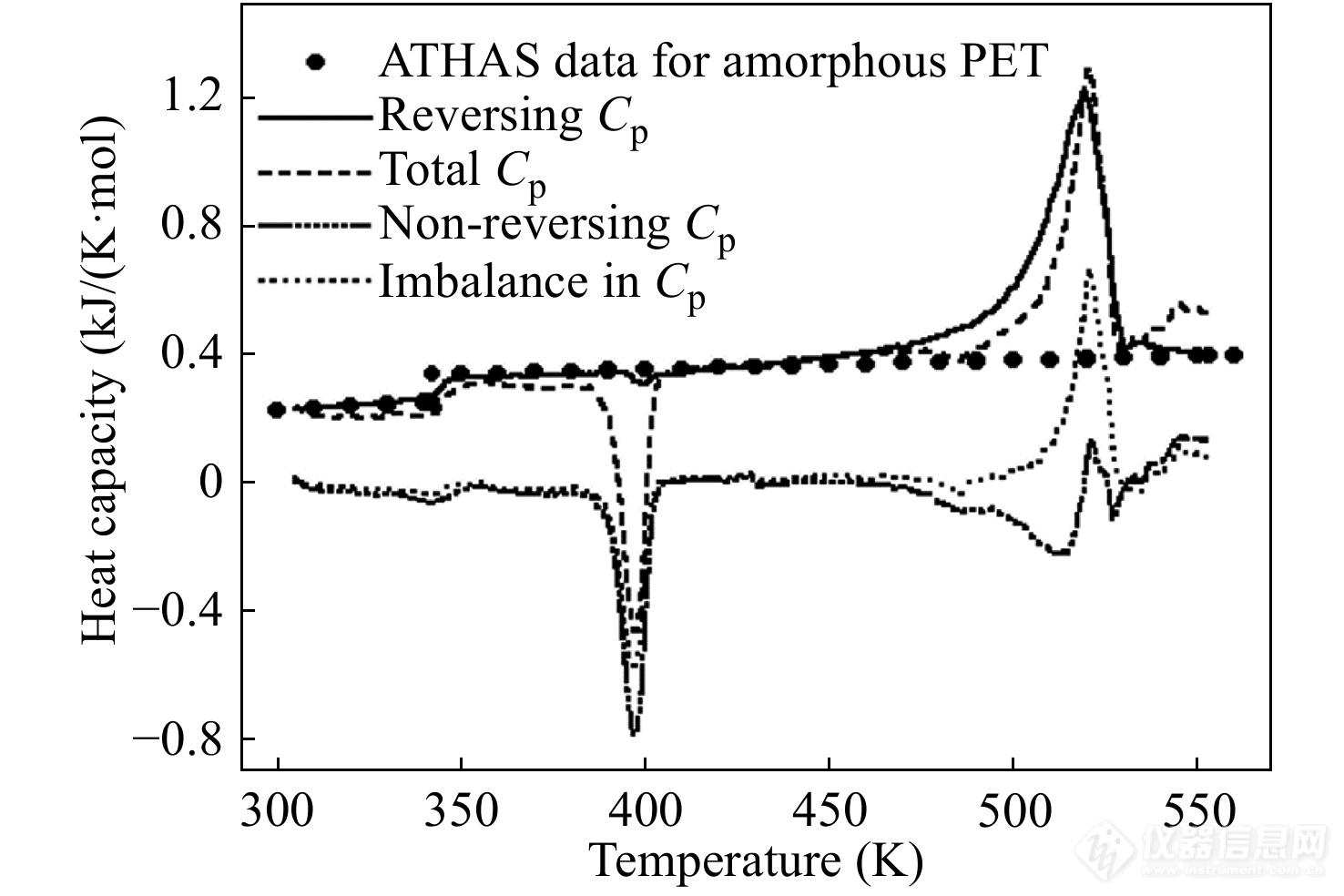
图 12
Figure 12. The heat capacity curves of poly(ethylene terephthalate) (PET) measured by sawtooth TMDSC with temperature profile of Fig. 11. The heat flow data is analyzed with the standard DSC method: reversing heat capacity from Eq. (26), total heat capacity from Eq. (25), non-reversing heat capacity from the difference between total and reversing heat capacity, and imbalance of heat capacity from Eq. (28). Also listed are the ATHAS data bank data for the heat capacity of amorphous PET. (Reprinted with permission from Ref.[35]; Copyright (2014) Springer Nature)
3.3.2 等温可逆热容
TMDSC的另一个重要应用是测量等温可逆热容. 传统DSC只能通过测量在一定温度梯度下的热流变化来测量热容,因此,传统DSC无法测量等温过程中的热容及其变化. 而TMDSC可以在基础升温速率为零的条件下,给样品施加一个调制的微小扰动速率,对样品进行准等温TMDSC实验,测量样品在等温过程中的热容及其变化.
图13是Wunderlich[36]对PET进行准等温TMDSC实验得到的比热容随温度变化的示意图. 图中较粗的实线代表了准等温实验测量得到的可逆比热容,较细的实线表示采用普通DSC在10 K·min−1的速率下测量得到的表观比热容,虚线表示理论计算得到的完全可逆的分子热振动比热容. 3条曲线在熔融峰区域以外的比热值基本一致. 而在熔融峰区域内,可逆比热值远小于表观比热值,这是因为标准DSC测量结果还包括了熔融相变潜热的释放. 另一方面,熔融峰区域的可逆比热仍高于基础热振动比热,这表明PET在熔程内出现了剩余热容,这部分剩余热容与半结晶高分子中大量存在的晶区与非晶区界面有关. 进一步研究发现,当升温速率较快时,剩余可逆热容会被抑制,由此推测剩余热容与晶体界面区的可逆熔融有关[37,38].
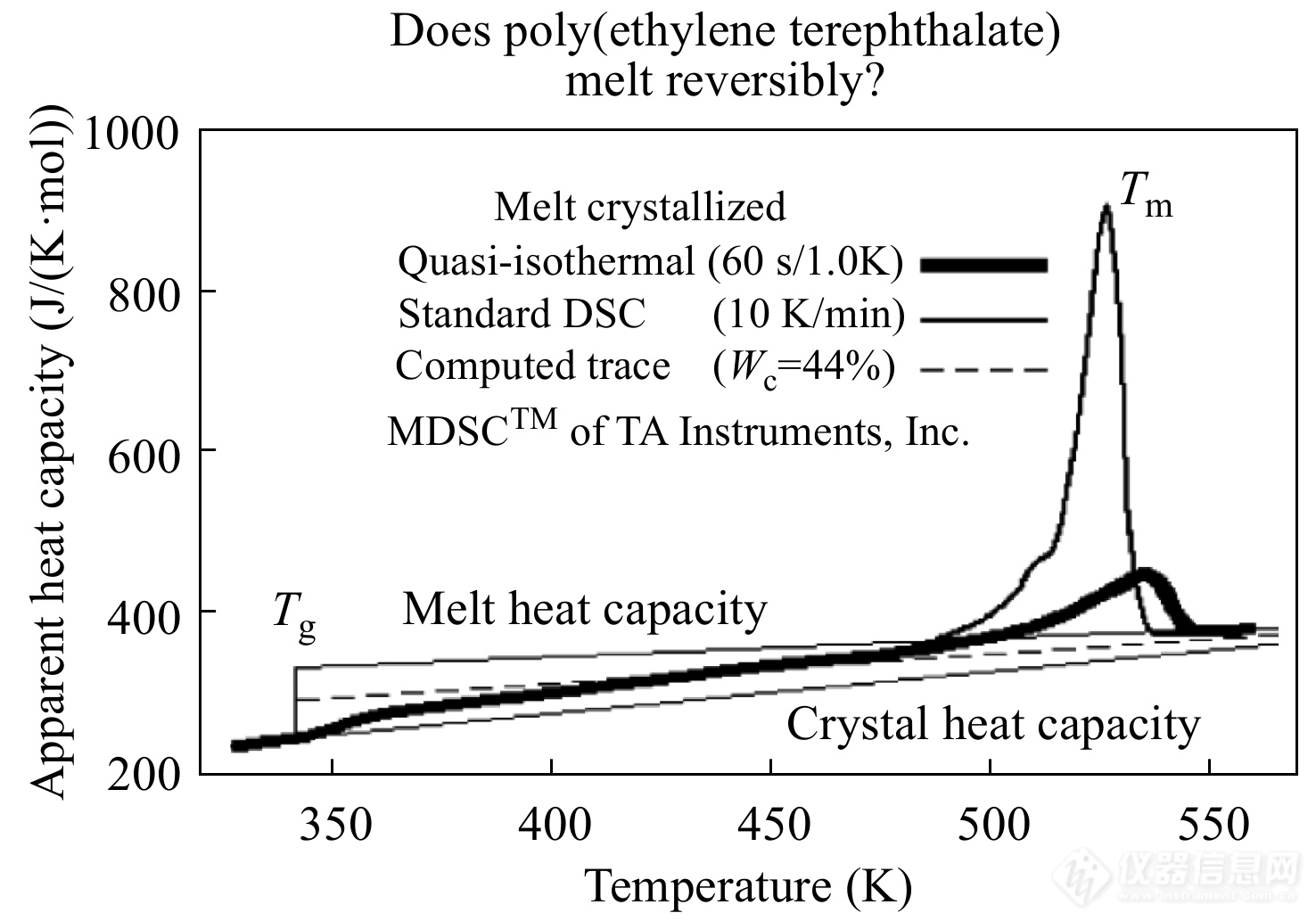
图 13
Figure 13. The apparent heat capacity curves of PET during the heating process after crystallized by cooling from the melt to 44% crystallinity. The standard DSC curve and TMDSC curve are separately with intermediate and heavy thickness. Also plotted are the data-bank information (thin line) and the computed heat capacity for the sample of 44% crystalline PET (broken line). (Reprinted with permission from Ref.[36]; Copyright (2014) Elsevier)
胡文兵等[39]进一步采用准等温TMDSC研究了几种链滑移能力不同的高分子在熔融温度范围内可逆热容的变化. 结果表明,链滑移能力较强的PE和PEO具有较大的可逆热容,而链滑移能力不强的PCL和PET测量得到的可逆热容较小,与熔体热容相近. 这种差别说明,剩余可逆热容是由发生在高分子片晶折叠端表面的可逆熔融所导致的,这种可逆熔融过程与分子链的链滑移能力密切相关. 作者由此提出了图14所示的折叠端表面的可逆熔化机制.
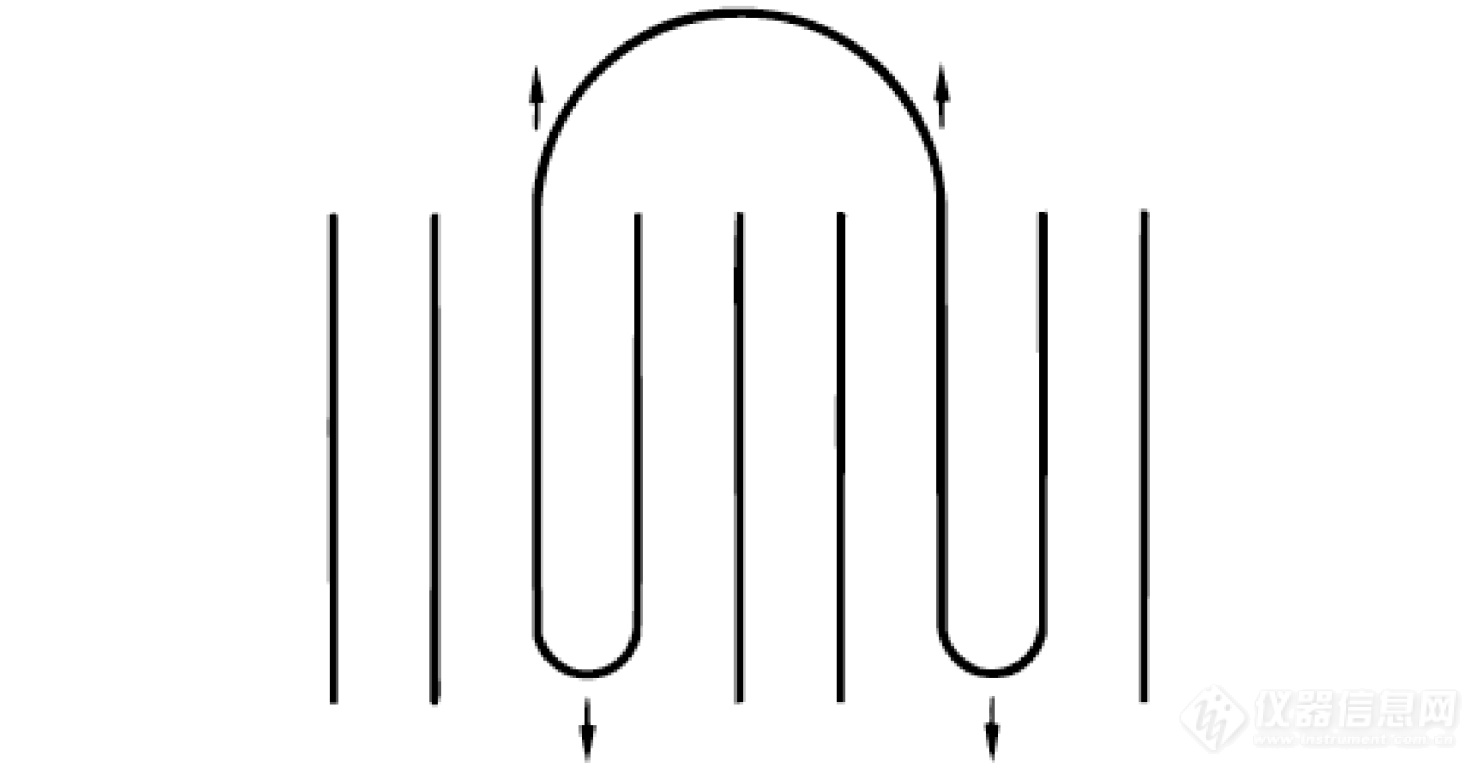
图 14
Figure 14. Illustration of reversible premelting on the fold-end surface of polymer lamellar crystals. There exists a local force balance between the recovery tendency of the stretched loops and the thickening tendency of the lamellar crystals (see arrows). (Reprinted with permission from Ref.[39]; Copyright (2014) American Chemical Society).
在高温区,为了满足表面环圈和纤毛的构象熵增大的需求,片晶折叠端表面的一部分链茎杆将通过滑移的方式抽出片晶,导致片晶的减薄,部分晶体发生熔融. 而在低温区,过冷度较高,结晶的热力学驱动力增强,在高温区部分熔融的片晶将通过链滑移进行晶区恢复,导致片晶增厚. 因此,随着温度的周期性变化,片晶折叠端表面出现可逆的熔融潜热释放,TMDSC信号上表现出超出分子热振动热容显著的剩余可逆热容.
江晓明等[40]采用TMDSC比较了α和β这2种不同晶型的iPP在高温下的可逆热容,并采用Monte Carlo分子模拟研究了上述调制过程. 结果如图15所示,2种晶型的iPP的可逆热容均随着调制频率的升高而降低,其中,链滑移能力较高的β晶型iPP具有更高的可逆热容,从而证明了链滑移能力在片晶折叠端表面的可逆熔融过程中的重要作用.
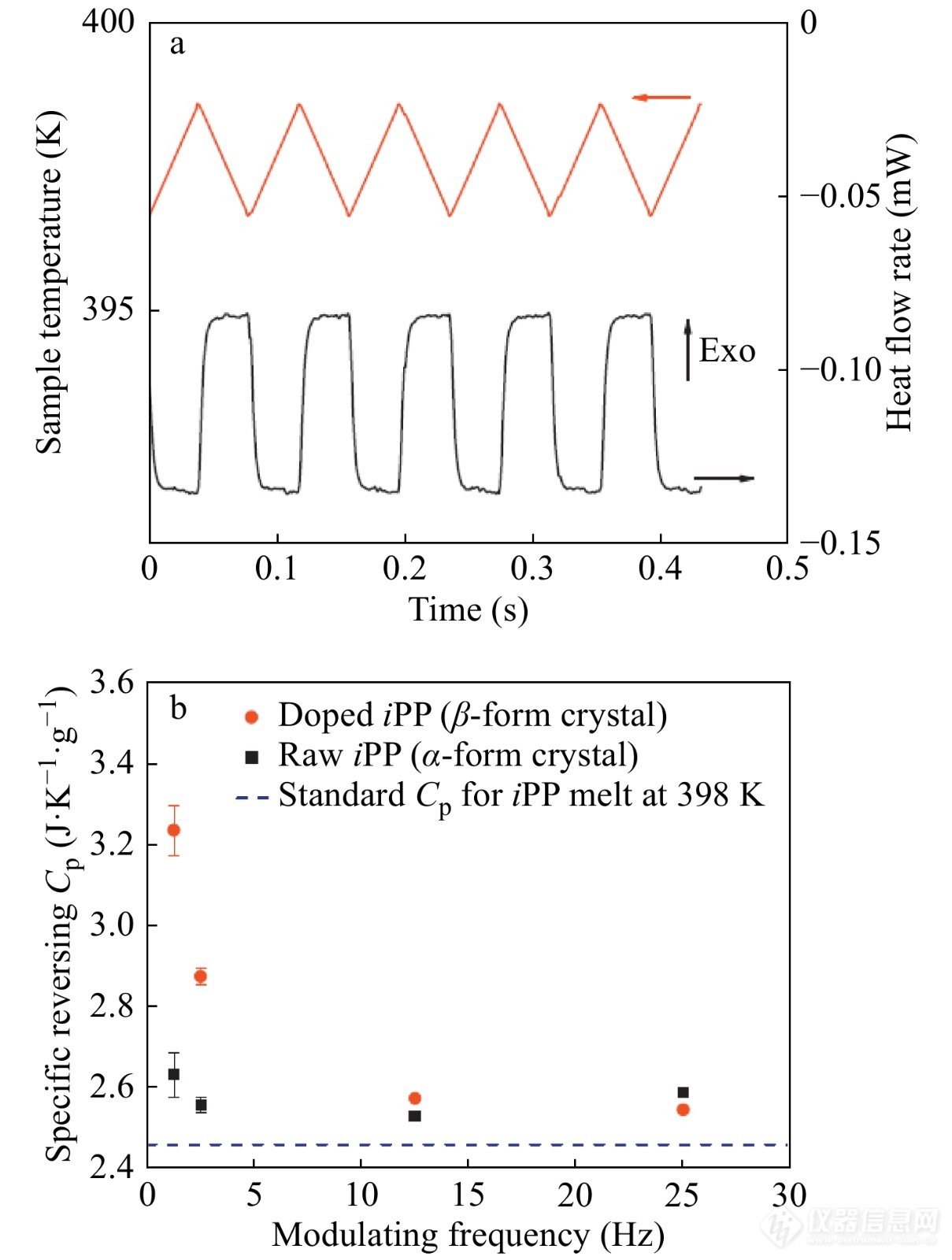
图 15
Figure 15. (a) The heat-flow rate curve (the black curve in the right axis) of the doped iPP as a response to the temperature-modulation program (the red curve in the left axis) with the frequency 12.5 Hz, the amplitude ±1 K and the baseline annealing temperature 398 K. (b) Frequency dependences of specific reversing heat capacities of raw and doped iPP samples measured by sawtooth TMDSC. The dashed line represents the standard specific vibrational heat capacity for iPP melt at 398 K that is cited from the literature [41]. (Reprinted with permission from Ref.[40]; Copyright (2014) Elsevier)
3.3.3 玻璃化转变
玻璃化转变常常与焓松弛、冷结晶等热效应重叠,TMDSC可以有效地区分玻璃化转变和其他热效应,从而准确测量玻璃化转变温度. 图16是采用TMDSC测量PS在353.15 K等温240 min后的升温热流曲线,左边的图包括了原始调制热流信号以及相应的总热流信号、可逆热流信号和不可逆热流信号. 将左图的纵坐标放大可得到右图,其中玻璃化转变为可逆热流信号,而焓松弛为不可逆热流信号,TMDSC可有效分离这2种热效应[42].
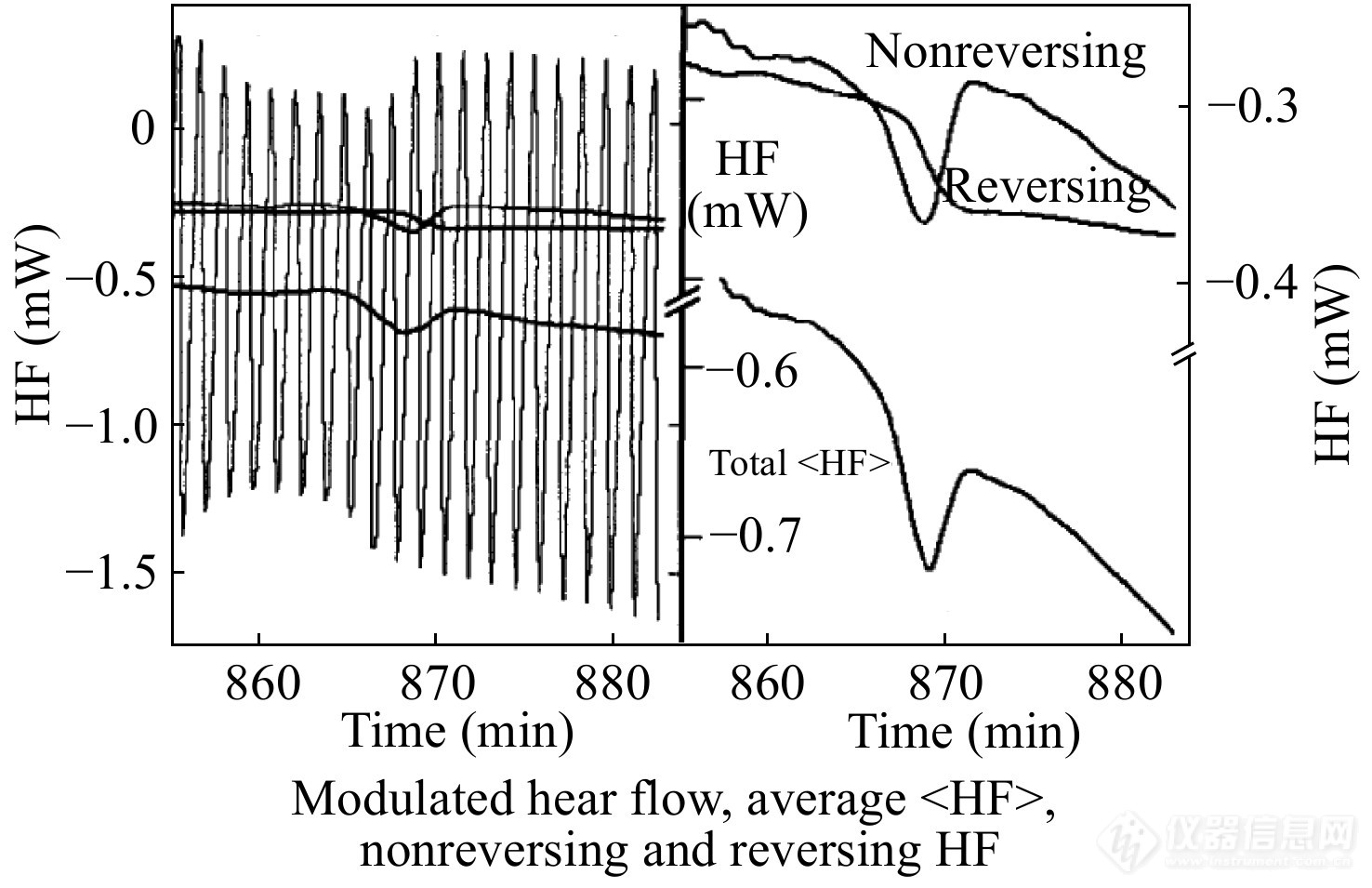
图 16
Figure 16. TMDSC measurement with the underlying heating rate 2 K·min−1, modulation period 80.5 s, and modulation amplitude 1.0 K for PS after annealing for 240 min at 353.15 K in order to separate the reversing and non-reversing contributions to the apparent heat capacity in the glass transition temperature region. Left figure: Modulated heat flow, the sliding averages, and the evaluated reversing and non-reversing heat capacities; Right figure: Expanded scale drawings of the three sliding averages. (Reprinted with permission from Ref.[42]; Copyright (2014) Elsevier)
玻璃化转变是一个动态变化过程,其热容变化具有频率依赖性. TMDSC能在2个时间尺度上测量玻璃化转变,包括较快的调制频率和较慢的平均升降温速率. 其中,调制热流信号测得的玻璃化转变温度与其热历史(最大升降温速率、退火温度等)无关,而只与调制频率有关,因此,TMDSC可以准确测量玻璃化转变过程中的热容变化的频率依赖性. 例如,图17是采用TMDSC测量PLA-H(含有16.4% D型旋光异构体的左旋聚乳酸PLLA)在不同调制频率下由373 K降温至283 K过程中的可逆比热容曲线. TMDSC的温度程序的参数为:基础降温速率为0.1 K·min−1,温度振幅AT为0.05~0.5 K,调制周期p为10~100 s. 在测试过程中,保持最大降温速率ATω不变,ATω=π/100,改变调制频率,ω=0.01~0.1 Hz,得到不同调制频率下的玻璃化转变温度,由此可计算出PLA-H在玻璃化转变区域的活化能[43].
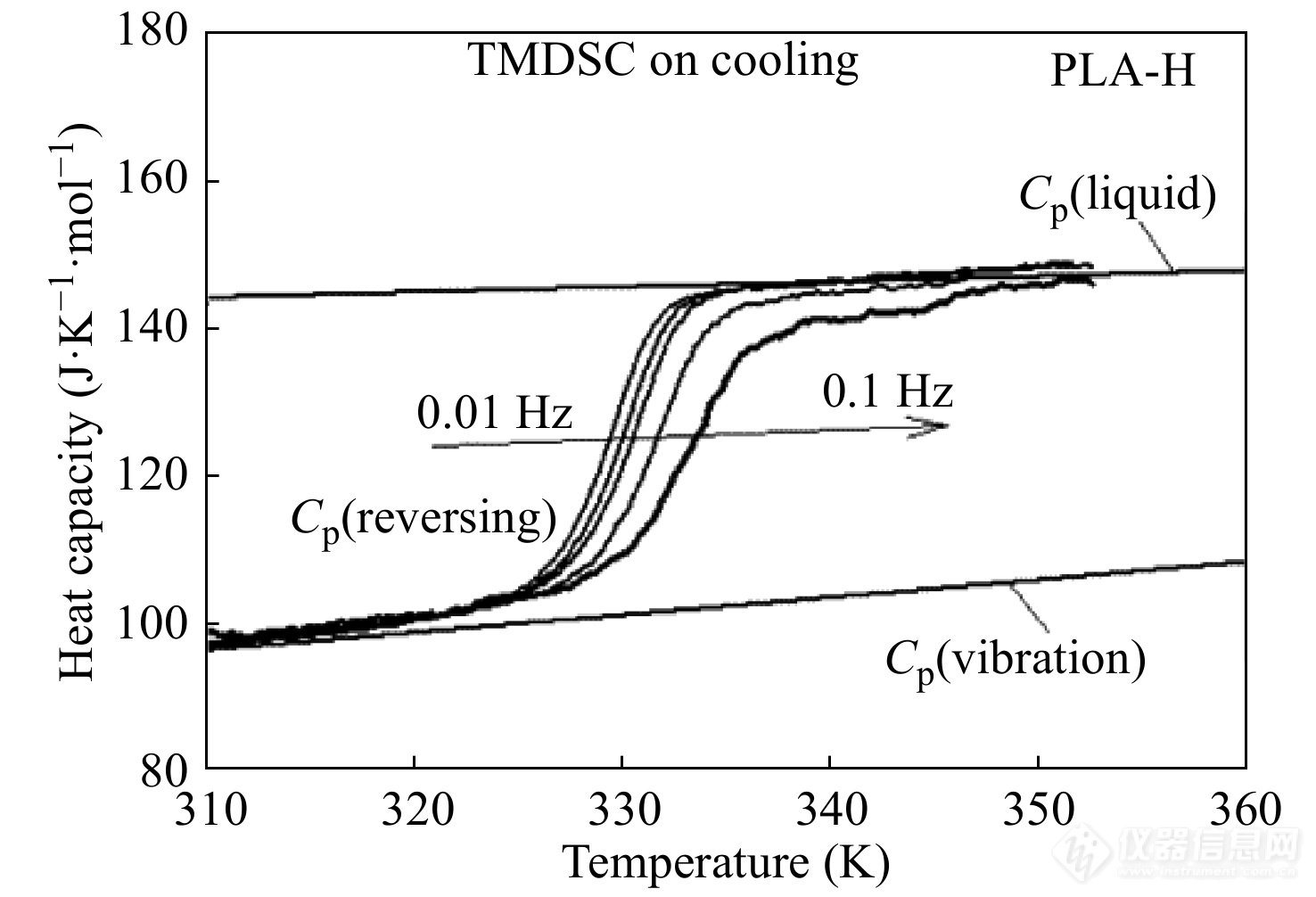
图 17
Figure 17. Specific reversing heat capacity curves of PLA-H cooled from 373 K to 283 K in TMDSC at different modulation frequencies. The underlying cooling rate is 0.1 K·min−1, and the maximum cooling rate ATω remains at π/100 with the modulation amplitude ranging from 0.05 K to 0.5 K and the modulation period ranging from 10 s to 100 s resulting in a wide range of modulation frequency from 0.01 Hz to 100 Hz. (Reprinted with permission from Ref.[43]; Copyright (2014) American Chemical Society)
4. 闪速示差扫描量热法
4.1 基本原理
20世纪60年代以来,DSC就已经成为了高分子材料研究领域尤其是高分子结晶学研究领域常用的实验研究手段. 然而,传统DSC的扫描速率比较小,一般在0.01~5 K s−1数量级范围内,阻碍了高分子结晶学领域研究的深入发展. 一方面,常规DSC无法抑制结晶速率较快的半结晶高分子样品在降温过程中的结晶成核以及在升温过程中的结构重组,从而限制了在较低温度区域内对高分子结晶成核行为的研究. 另一方面,由于实际生产加工过程中的降温速率极高,例如吹塑和注塑的降温速率可达到100~1000 K·s−1,因此常规DSC无法模拟高分子在实际生产加工过程中的结晶环境[44,45].
DSC的升降温速率以及温度控制的灵敏度亟待提高. 然而,较快的升温速率会导致样品内部出现较大的温度梯度,热滞后影响了热流信号的可重复性和准确性,
|
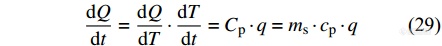
依照DSC的热流信号公式(29),在提高扫描速率q的基础之上减小样品质量m,既可以保证热流信号的灵敏度,同时也减轻了较大质量的样品在快速扫描过程中的热滞后效应. 因此,DSC开始朝着微型化、高速化发展,闪速示差扫描量热仪(FSC)由此诞生. FSC采用氮化硅芯片传感器替代传统DSC的坩埚,将样品质量由原来的毫克级别减小到了纳克级别,有效避免了样品内部的热滞后,并能通过芯片传感器进行温度的控制和热量的补偿,实现了快速的升降温扫描,大大拓展了高分子表征的时间和空间灵敏度.
FSC技术得益于20世纪90年代氮化硅薄膜和微机电系统(micro electro mechanical systems, MEMS)技术的发展. 1994年,Hellman等[46]首次制备出无定形氮化硅薄膜传感器,并基于该传感器研制出附加热容约为4×10−6 J·K−1的交流式薄膜微量热仪. 微小的附加热容能有效避免热滞后,有利于扫描速率的提升. 2004年,Allen等[47,48]基于氮化硅薄膜传感器研发出升温速率可达到105 K·s−1的薄膜示差扫描量热法(thin film differential scanning calorimetry, TDSC). 然而,TDSC采用了真空环境制备准绝热条件,导致仪器散热困难,无法实现快速的降温扫描. 同年,Schick等[49,50]采用商用热导器件TCG-3880 (Xensor Intergrations, NL)优化功率补偿型薄膜芯片量热仪,使用氮气、氦气等气氛,将非绝热环境下可控的降温速率提高到106 K·s−1. 2005年,唐祯安[51]研发出加热速率可达2×105 K·s−1的微量热仪. 近年来,周东山设计出冷热台型高速扫描量热仪,可将高速扫描量热技术与显微红外光谱、拉曼光谱、X射线衍射以及原子力显微镜等微结构光学表征技术连用[52],能够捕捉结晶性高分子及液晶小分子的亚稳态结构,更准确地表征高分子多相结构转变、共混及共聚物中结晶相空间结构、以及纳微米受限态下高分子的成核结晶动力学[53~55].
随着氮化硅薄膜技术的发展,商业化的快速扫描量热仪的研发也不断取得进展. 从2003年起,Xensor、Anatech、SciTe三家公司开始合作研发商业化快速扫描量热仪,并在随后开发出XI-400型陶瓷基板芯片传感器(UFS1). 2010年,瑞士Mettler-Toledo公司(国内称梅特勒公司)[56]基于UFS1芯片传感器技术成功开发出第一代商业化功率补偿型快速扫描量热仪Flash DSC 1.
图18是Flash DSC1设备的示意图. 左上角展示了Flash DSC1的仪器主机及其配备的显微镜. 该显微镜由德国莱卡公司生产,放大倍数为2000,主要用于辅助样品制备和观察芯片传感器的状况. 右上角是该仪器配备的XI-400型陶瓷(UFS1)芯片传感器,传感器背部有16个接触位点,可与主机芯片装载台上的接线柱相连接,实现温度控制、热量补偿和数据控制,UFS1是Flash DSC1实现快速升降温速率以及精准控温的关键性设备[57]. 左下角的图片展示了安装好传感器并盖上盖板的装载台. 右下角展示的是在光学显微镜下的样品池或参比池,其中黑色圆形是直径为500 μm的有效加热区. 该仪器配备了德国Huber TC100机械制冷机,可实现在−100~450 °C温度范围内的快速升降温. Flash DSC1的升温速率范围在0.5~40000 K·s−1,降温速率的范围在0.1~4000 K·s−1. 目前瑞士梅特勒公司已推出降温速率高一个数量级、升温范围高达1000 °C的第二代设备Flash DSC2+.

图 18
Figure 18. The photographs of Flash DSC1 apparatus. Top left: Flash DSC1; Top right: the unloaded chip sensor UFS1; Bottom left: the sample transfer; Bottom right: the membrane of the sample or reference cell on sensor. (Reprinted with permission from METTLER-TOLEDO Company)
近年来,随着商业Flash DSC设备的不断完善和发展,FSC在PCL[58,59]、iPP[60~62]以及iPB[63]等多种高分子材料的结晶、成核以及熔融动力学等表征中得到了越来越广泛的应用.
与传统DSC相比,FSC的时间常数由秒降到了毫秒级别,大大缩短了实验的观测窗口,可在纳米尺度上考察分子链的运动过程,大大促进了对高分子亚稳态结构相转变动力学行为的研究. 同时,FSC将样品量由原来的毫克减小到了纳克级别,将DSC技术的研究范围拓展到了微纳米高分子材料体系[64].
4.2 实验技巧
4.2.1 样品制备
FSC中的样品制备过程与传统DSC有较大的区别. 通常Flash DSC的样品质量为5 ng到几微克. 较少的样品量有利于提高样品与传感器之间的热接触,减小热滞后效应,得到更尖锐的信号峰和更准确的测量结果. 然而,样品过少会导致热流信号灵敏度过低,还可能带来尺寸效应. 因此,可以根据温度程序的扫描速率选择合适的样品量. 当扫描速率大于1000 K·s−1时,样品质量小于100 ng;当扫描速率低于20 K·s−1时,为了保证热流信号的灵敏度,样品质量可取几百纳克[65].
实验过程中样品直接放置在FSC的传感器上. 可以将芯片传感器取下来在外部进行样品制备,例如旋涂、蒸发沉积等前处理,也可以借助仪器自身配备的显微镜直接切割样品. 当初始样品是薄膜、挤出粒子、粉末颗粒这类体积较大的物质时,在显微镜下用手术刀将初始样品切割成厚度小于10 μm的薄片,然后将样品转移到干净的载玻片上,进一步将样品切割成面积为50 μm×50 μm的薄片,然后用自然带有尖端的细毛提取样品将其转移至位于芯片样品池中央的圆形加热区[65]. 以1 K·s−1的速率对样品进行预熔,使样品与芯片表面具有良好的热接触,同时降低样品对芯片传感器的机械应力. 当样品与传感器的热接触效果不好时,在不影响测试结果的前提下,可在上样之前在传感器表面涂一薄层硅油作为热接触媒介. 除了提高热接触,硅油还可用于降低样品的机械应力,测试初次升温扫描的结果,防止样品在升温过程中弹出加热区,提高芯片传感器的重复利用次数等.
4.2.2 样品质量
FSC的样品量过小,无法采用天平直接测量样品的质量,通常需要根据样品的性质进行估算. 较为粗糙的方法是根据样品的尺寸和密度进行估算[66]. 较为准确的方法是利用样品的热性质,包括热容[67]、熔融焓[68]以及玻璃化转变台阶的热容差[69]来计算样品质量,可根据样品的特点选择不同的热性质进行质量测量.
例如,依照样品在熔融状态下的热容计算样品质量的公式为
|
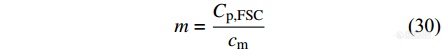
其中,[Math Processing Error]Cp,FSC是采用Flash DSC1测量得到的样品在某一温度范围内的平均表观热容. [Math Processing Error]cm是样品的比热容,可通过常规DSC准确测量一定质量的样品在该温度范围内的热容,由热容与质量的比值得到该样品的比热容,也可以通过数据库查找标准比热容值.
同理可得到熔融焓法计算样品的公式
|
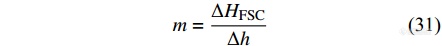
其中,[Math Processing Error]ΔHFSC是Flash DSC1测量得到的样品的熔融焓. [Math Processing Error]Δh为单位质量样品的熔融焓,一般采用传统DSC对具有相同结晶条件的样品进行测量得到.
利用样品的玻璃化转变台阶计算样品质量的公式为
|
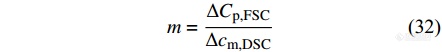
其中,[Math Processing Error]ΔCp,FSC是采用Flash DSC快速降温得到的完全无定形态非晶样品的玻璃化转变台阶处的热容变化值. 采用浸入液氮等外部方法制备无定形态样品,然后放入常规DSC中测量,即可得到该样品在玻璃化转变处的比热容变化值[Math Processing Error]Δcm,DSC.
4.2.3 临界条件
FSC技术的一大优势是通过调节降温速度获得不同相态结构的化合物,包括无定形态、介晶态以及结晶态. 因此,在进行温度程序设计之前需要了解制备不同相态结构样品的临界升降温速率,包括消除热历史的临界温度以及临界扫描速率的测试.
消除热历史实验指的是将样品升温至足够高的温度等温一段时间以消除熔体中残留的晶体或晶核,避免记忆效应. 消除热历史的温度一般在熔点和分解温度之间,温度过高会导致样品发生热降解. C66是82%(摩尔分数)PA6与18%的PA66组成的无规共聚物. 采用Flash DSC1测定C66样品消除热历史所需的临界温度时,先将样品加热至不同的温度等温0.2 s消除热历史,然后以−10 K·s−1的速率冷却至−100 °C,最后以3000 K·s−1的速率升温至250 °C,得到如图19所示的加热曲线. 当消除热历史温度高于170 °C时,熔融峰相互重叠,表明高温等温已经完全消除了样品中的热历史,得到C66样品消除热历史的临界温度为170 °C. 由于均聚物PA6的平衡熔点为250 °C,实验中可选择270 °C等温0.2 s作为消除热历史的温度程序.
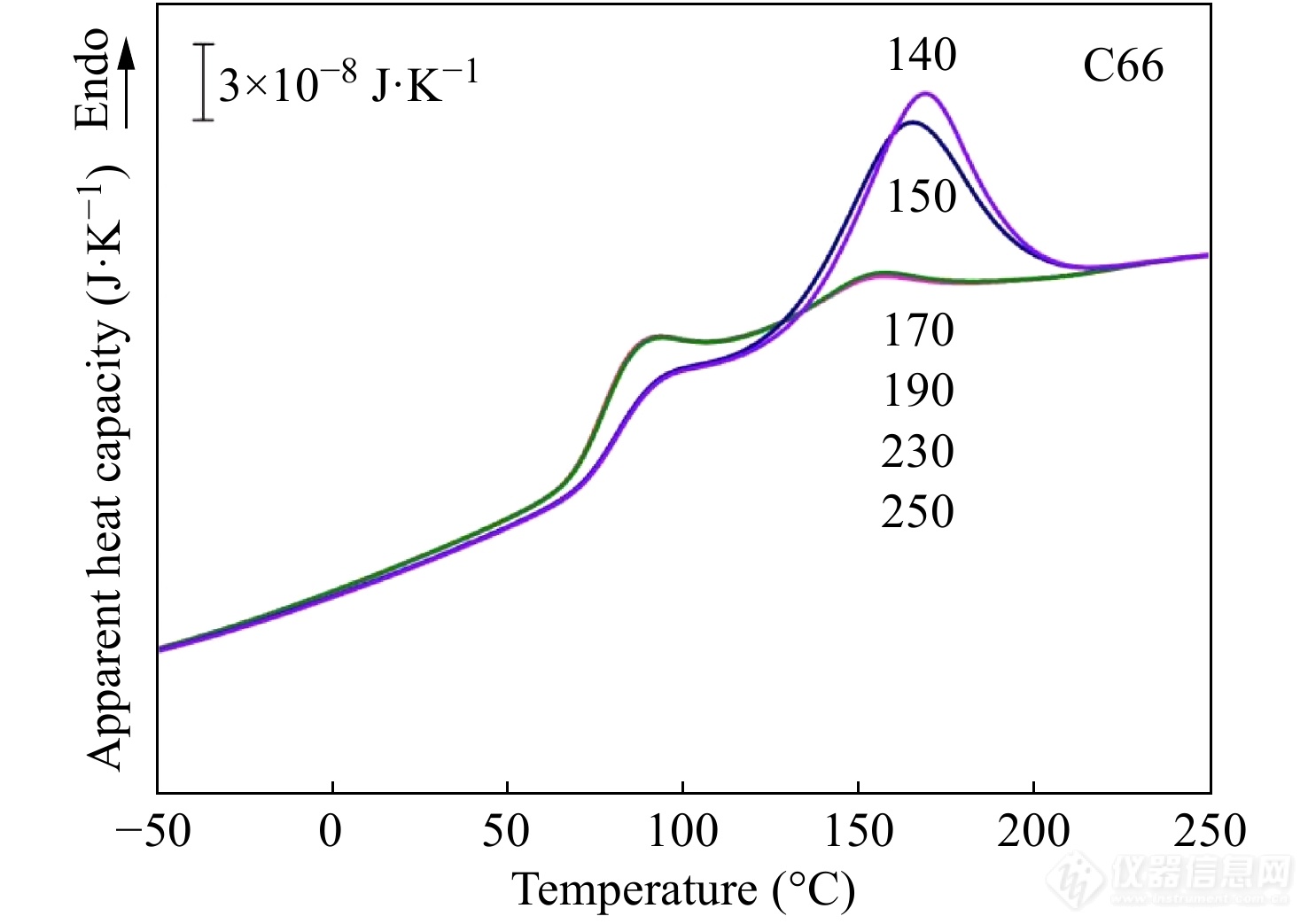
图 19
Figure 19. Apparent heat capacity curves of C66 samples obtained on heating at 3000 K·s−1 after cooled at −10 K·s−1 from a stay of 0.2 s at different erasing temperatures ranging from 180 ℃ to 210 ℃ (Reprinted with permission from Ref.[70]; Copyright (2014) Elsevier)
临界扫描速率包括临界升温速率和临界降温速率,它是结晶动力学研究的一个重要临界条件. 临界降温速率指的是恰好能够抑制样品在降温过程中发生结晶的临界速率. 图20是iPP样品(V30G)在消除热历史之后以不同的速率降温得到的降温过程中的热容曲线. 当降温速率超过500 K·s−1时,结晶峰消失,说明样品的临界降温速率为500 K·s−1.
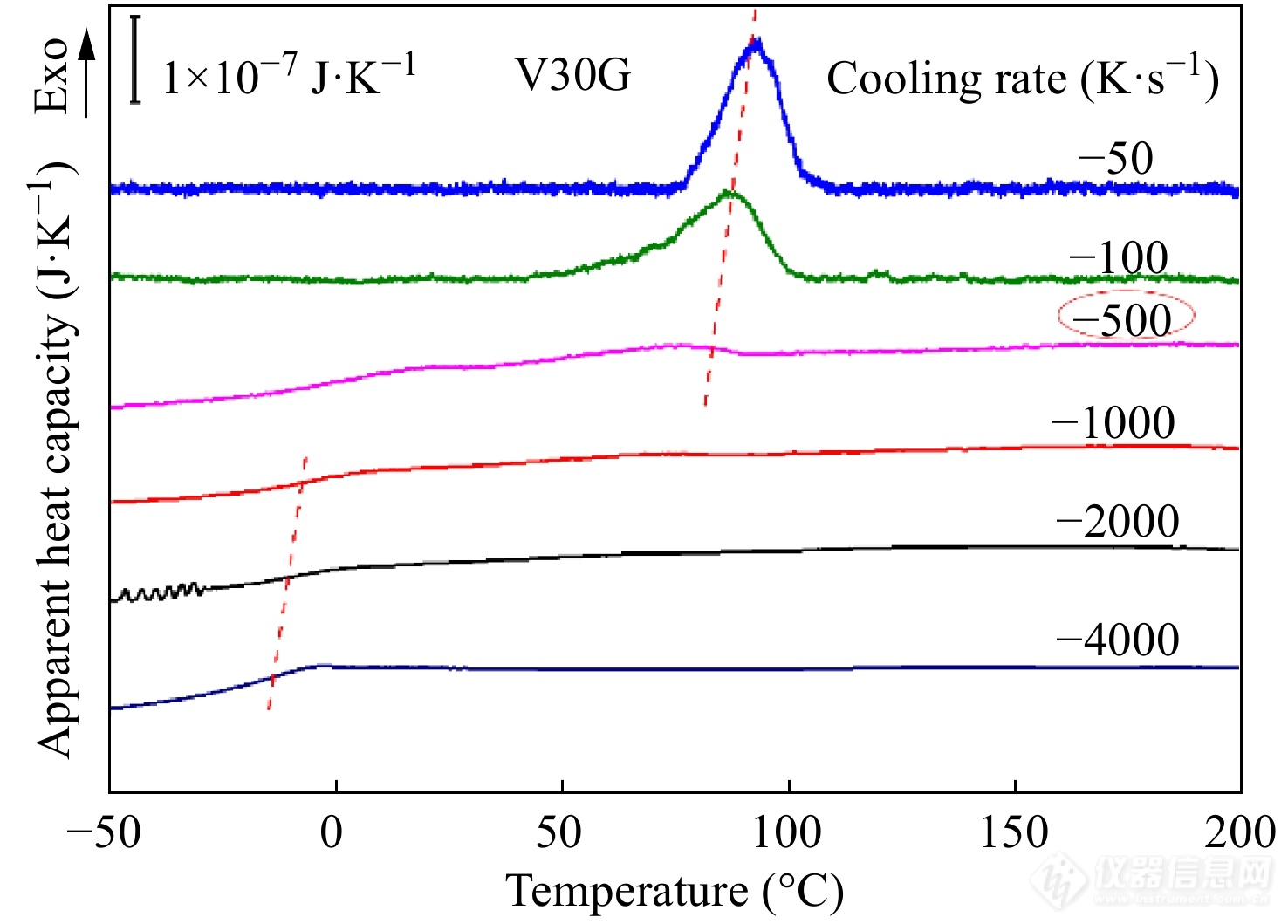
图 20
Figure 20. Apparent heat capacity curves of V30G sample obtained on cooling at various rates as labeled (Reprinted with permission from Ref.[60]; Copyright (2014) Springer Nature).
临界升温速率指的是恰好能够抑制样品在升温过程中出现冷结晶的临界速率. 将上述V30G样品以超过临界降温速率冷却至玻璃化转变温度以下,然后以不同速率升温至熔点以上,得到如图21所示的升温过程中的表观热容曲线. 随着升温速率逐渐增大,升温曲线上的冷结晶峰和熔融峰变得越来越微弱. 当升温速率达到30000 K·s−1时,冷结晶峰消失,表明V30G样品的临界升温速率为30000 K·s−1.
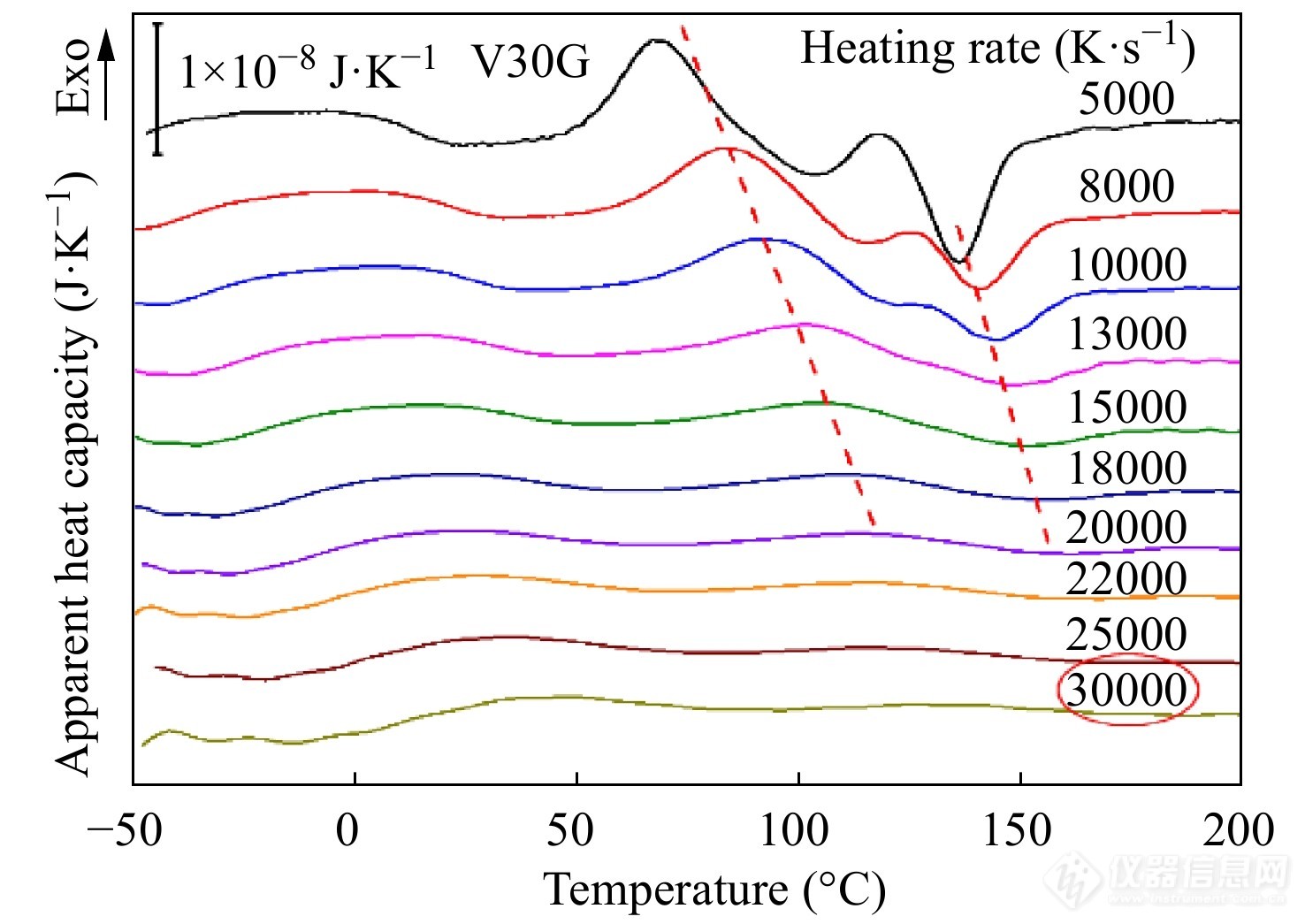
图 21
Figure 21. Apparent heat capacity curves of V30G sample obtained on heating at various rates as labeled (Reprinted with permission from Ref.[60]; Copyright (2014) Springer Nature)
得到上述临界条件之后就可以进一步对高分子相转变动力学行为进行研究,包括测量样品的总结晶动力学、结晶成核动力学、晶体熔化动力学、晶体退火动力学等.
4.3 应用举例
4.3.1 等温总结晶动力学
高分子结晶动力学行为是影响高分子产品的生产效率和产品性能的重要因素. 高分子总结晶动力学由晶体初级成核所控制. 根据经典成核理论,在高温区,高分子成核速率主要由临界成核自由能位垒所控制,而在低温区则由分子短程扩散活化能位垒所主导. 由于临界成核自由能位垒随着温度的升高而升高,而扩散活化能位垒随着温度的升高而降低,因此,高分子结晶速率对结晶温度的依赖性关系曲线呈抛物线形,其最快的结晶速率在玻璃化转变温度和熔点之间. 对于结晶速率较快的高分子,传统DSC的降温速率无法抑制它在高温区的结晶,从而对较低温度范围内的结晶动力学研究产生影响. 因此,传统DSC的结晶动力学研究只能局限在低过冷度的高温结晶区域. 而Flash DSC能抑制除了PTFE和PE以外大多数高分子在整个温度范围内的结晶,大大推进了对于低温区高分子结晶动力学行为的研究[71,72].
何裕成等[73]采用Flash DSC1对热力学条件相近的尼龙6 (PA6)和聚酮(PK)在全温度范围内的结晶动力学行为进行了对比,得到如图22所示的结晶动力学曲线. 在低温区,PA的分子层之间较强的氢键作用及其较高的玻璃化转变温度,削弱了PA的分子链运动能力,导致其结晶速率较慢. 而在高温区,PA中层状分布的氢键作用大大降低了层间的表面自由能,使得成核自由能位垒降低,大大加快了PA的结晶速率.
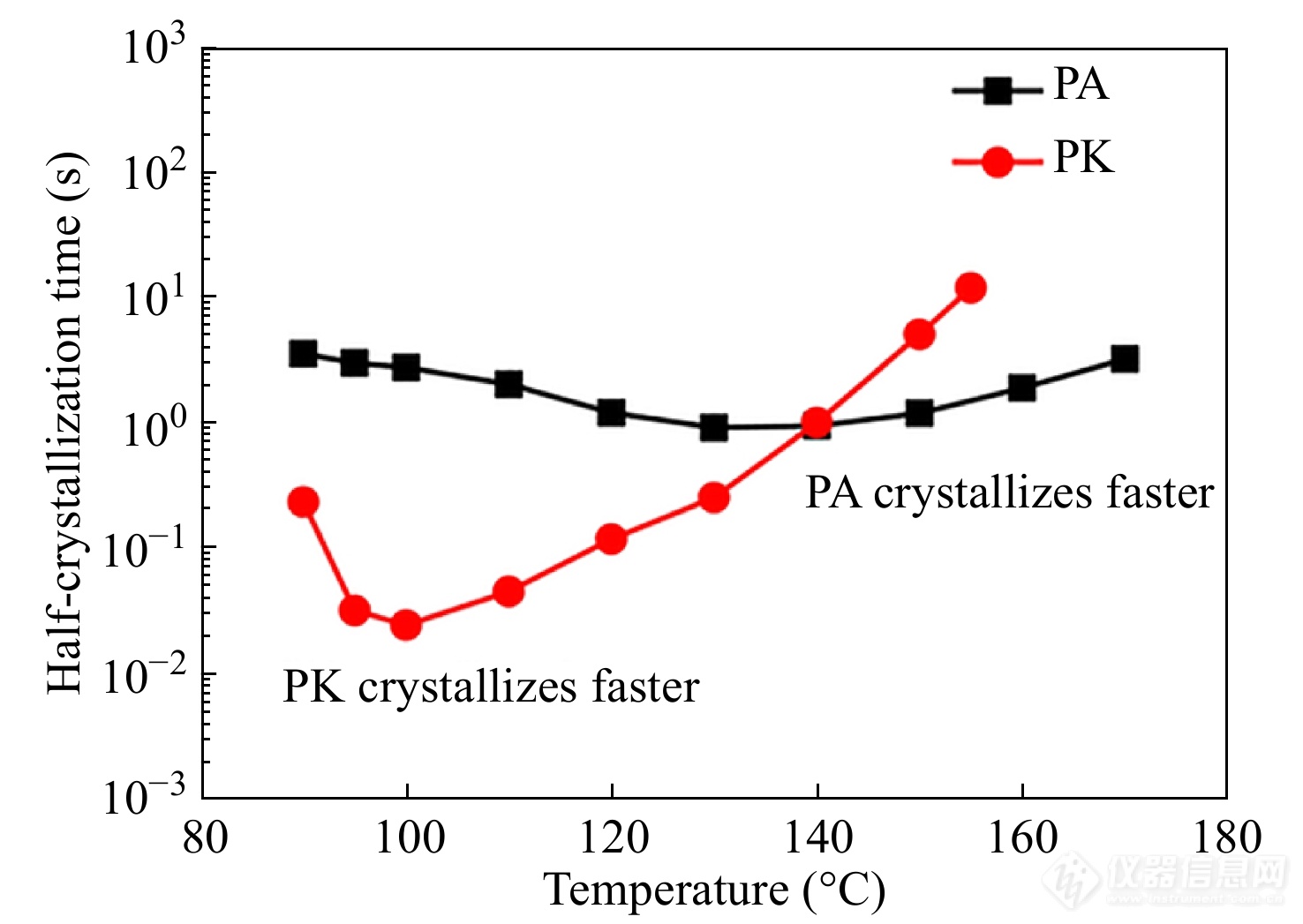
图 22
Figure 22. Comparison of temperature dependence of crystallization half-times of PA and PK during isothermal crystallization process at various crystallization temperatures (Reprinted with permission from Ref.[73]; Copyright (2014) John Wiley and Sons)
上述结果表明,氢键结构对聚酰胺的结晶动力学行为具有重要影响. 此外,聚酰胺的氢键结构与蛋白质的二级结构β折叠十分相似,对聚酰胺的氢键结构的研究有助于理解蛋白质β折叠的微观机制[74]. 因此,李小恒等[75]进一步采用Flash DSC1比较了6种聚酰胺(PA46, PA66, PA610, PA612, PA1012, PA12)在整个温度范围内的等温结晶动力学行为. 图23展示了不同聚酰胺样品的结晶动力学曲线. 其中,PA46的高氢键密度有利于提高高温区的热力学驱动力,加快结晶速率. 而PA10和PA12的低氢键密度有利于加快低温区的短程扩散,导致其较快的结晶速率. 此外,聚酰胺的半结晶时间-等温温度曲线呈现双峰型分布,表明了聚酰胺的成核方式由高温区的异相成核转变为低温区的均相成核,且该转变温度随氢键密度的改变而改变.
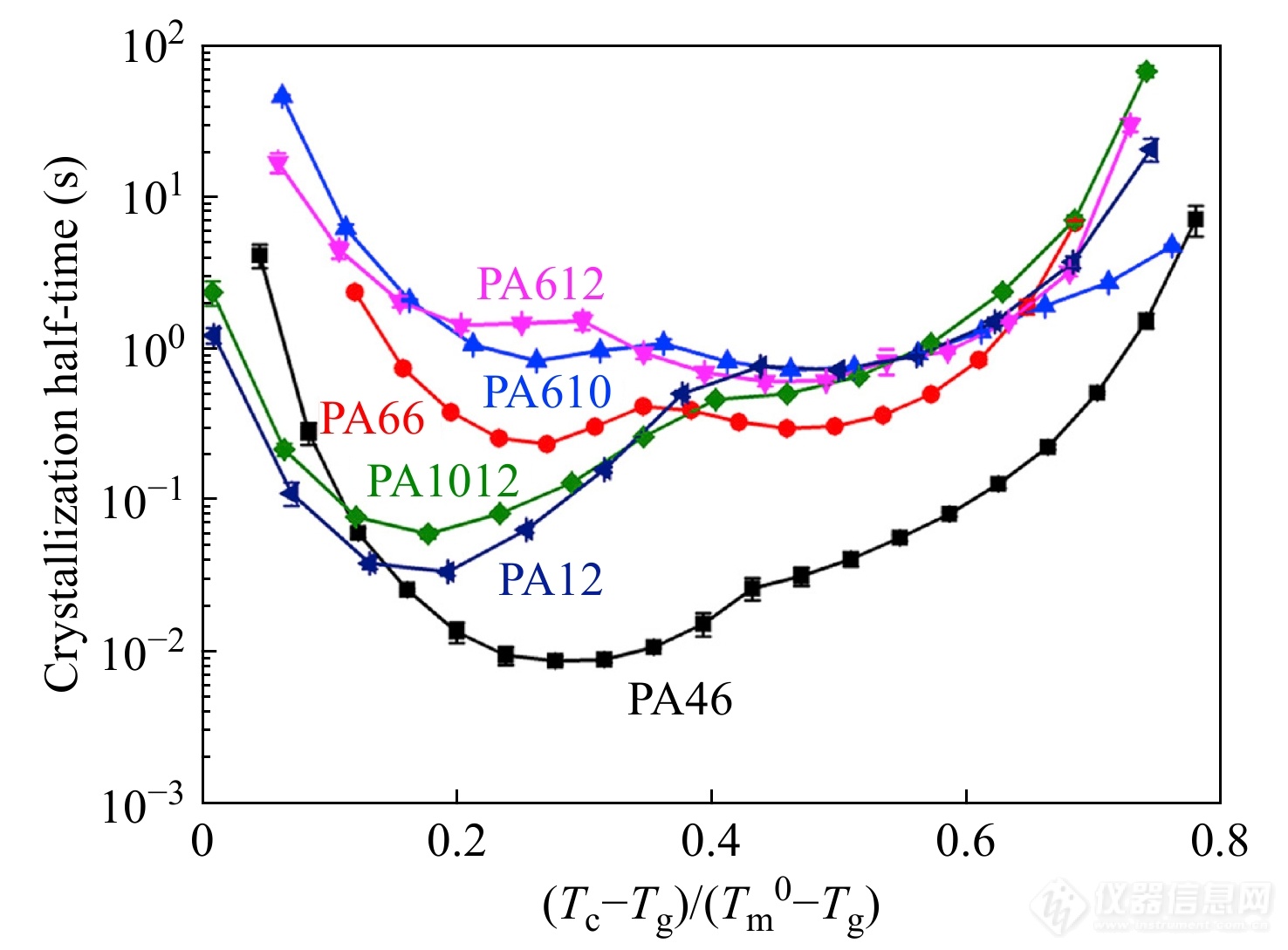
图 23
Figure 23. Summary of temperature dependence of crystallization half-times of PA46, PA66, PA610, PA612, PA1012 and PA12 during isothermal crystallization processes at various temperatures (Reprinted with permission from Ref.[75]; Copyright (2014) Elsevier)
4.3.2 不可逆熔融转变
高分子片晶在熔化的过程中伴随着熔融重结晶等结构重组优化过程的竞争,也就是所谓的非零熵熔融(non-zero-entropy-producing melting, non-ZEP melting). 当升温速率足够快时,所有的退火行为都将被抑制,此时观察到的熔融行为就反映了原始晶体自身的熔融行为,被称为零熵熔融(zero-entropy-producing melting, ZEP melting). 采用Flash DSC对高分子样品进行快速升温可以在某种程度上抑制亚稳态晶体在熔化过程中的结构优化,表征发生在高分子晶体侧表面的不可逆熔化动力学.
Toda等[76,77]研究了PET、iPP和PCL的片晶熔化动力学,首次发现了过热度Tm−Tc与升温速率R之间存在指数标度关系. 进一步研究发现这种特征的标度关系可能与晶体不可逆熔化的动力学机制有关. 高欢欢等[78]结合Flash DSC1和Monte Carlo分子模拟研究了由α晶型和β晶型iPP这2种化学结构相同但链滑移能力不同的高分子晶体在较宽的动态扫描速率范围内的过热度与升温速率的标度关系. 结果图24所示,该指数标度关系与iPP分子链在不同尺度上的分子链滑移以及分子内成核和片晶侧表面的粗糙化生长有关.
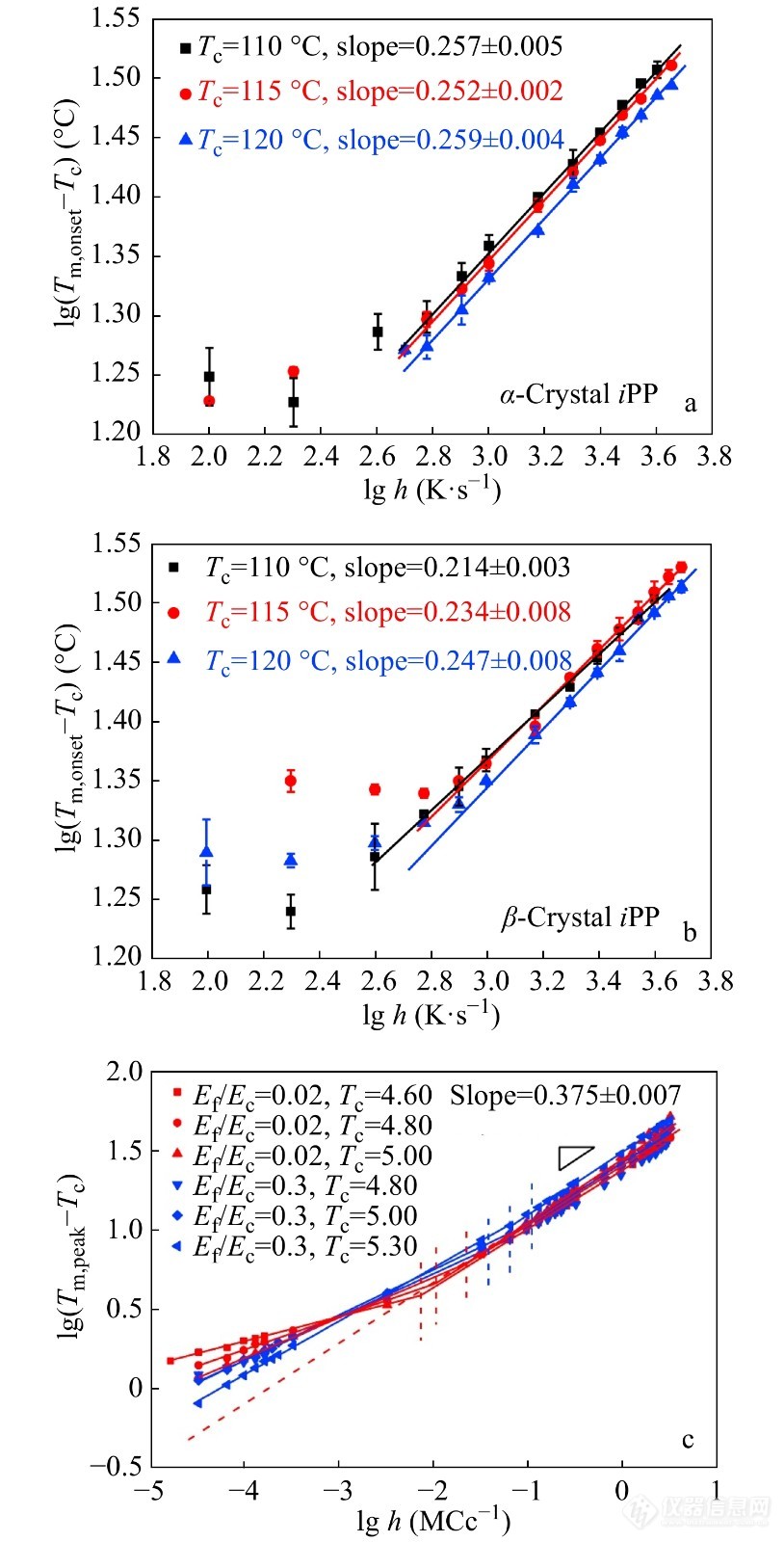
图 24
Figure 24. (a, b) FSC measurement of power law relationships between apparent superheating Tm,onset−Tc and heating rates h for α-crystals and β-crystals of iPP prepared at three crystallization temperatures Tc as labeled. (c) Mote Carlo simulations of power law relationship between apparent superheating Tm,onset−Tc and heating rates h for lamellar iPP crystals with different chain mobility characterized by Ef/Ec and different crystallization temperatures Tc as labeled (Reprinted with permission from Ref.[78]; Copyright (2014) Elsevier)
此外,采用FSC对聚合物进行快速升温,可避免聚合物的熔化和降解,从而得以研究高分子亚稳态结构的动力学变化过程. Monnier等[79]采用FSC以10000 K·s−1的速率加热吸附在固体表面的聚合物层,在较小的时间窗口内避免了样品的降解,直接观察到聚合物熔体在固体表面的解吸附现象. 实验结果表明,解吸附焓变与退火温度无关,吸附/解吸附是类似于结晶/熔融的一级热力学转变.
4.3.3 与其他表征技术连用
前面已经介绍到FSC技术可与其他表征技术连用来表征高分子材料[52~55]. FSC技术还可与X射线衍射[80],原子力显微镜[81~83]、偏光显微镜(polarized light optical microscopy, POM)[84]等多种分析仪器实时连用,进一步获得晶体的形态及微观结构的变化信息. 吕瑞华等[85]结合了FSC以及AFM研究了左旋聚乳酸(PLLA)的α'-α晶型转变机理. 图25(a)是左旋聚乳酸在152 °C等温退火不同时间的熔融曲线图. 红色曲线代表了α'晶,蓝色曲线为α晶. 由图可知,随着退火时间的增加,左旋聚乳酸晶体中出现了连续的晶体完善与不连续的熔融重结晶过程的竞争. 图25(b)和25(c)分别为初始结晶晶体和高温退火后的晶体的AFM图. 相较于初始结晶晶体,退火后的球晶尺寸更大,且晶核数量减少. 因此,PLLA在高温处的α'-α晶型转变机理是非连续的熔融重结晶过程.
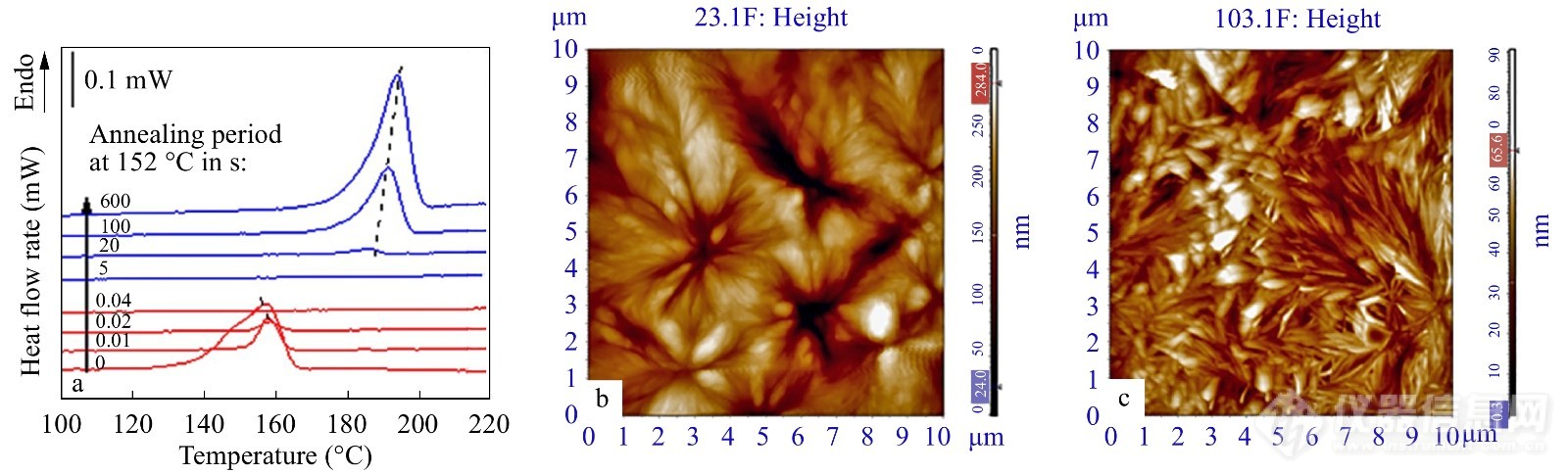
图 25
Figure 25. (a) Heat flow curves of PLLA crystals after annealing at 152 °C for various periods from 0 s to 600 s; (b) AFM height image of nascent PLLA crystals; (c) AFM height image of PLLA after annealed at 152 °C for 1000 s (Reprinted with permission from Ref.[85]; Copyright (2014) Elsevier)
4.3.4 玻璃化转变
FSC具有极宽的动态扫描速率范围,可用于制备各种不同的玻璃态结构. Schawe等[86]采用Flash DSC2+以不同降温速率将金属玻璃Au49Ag5.5Pd2.3Cu26.9Si16.3由熔体淬火至玻璃化温度以下,得到了2种不同的玻璃态结构:在中等降温速率下形成的自掺杂玻璃态结构(Self-doped glass, SDG)以及在较高降温速率下形成的化学均质玻璃态结构(chemically homogeneous glass, CHG). 对这2种新型玻璃态结构的研究有助于检验现有玻璃化转变理论的普适性,优化金属玻璃的生产加工条件.
FSC还可用于研究玻璃化转变在微纳米尺度上的受限效应. Monnier等[87]采用FSC以0.1~1000 K·s−1的不同降温速率将聚(对叔丁基苯乙烯)(poly-(4-tert-butylstyrene), PtBS)冷却至玻璃化转变温度以下,研究样品尺寸和降温速率对玻璃态结构的影响. 结果如图26所示,随着降温速率以及样品尺寸的降低,虚拟温度减小到远远低于本体的玻璃化温度,样品松弛到平衡态所需的时间也随之大大缩短.
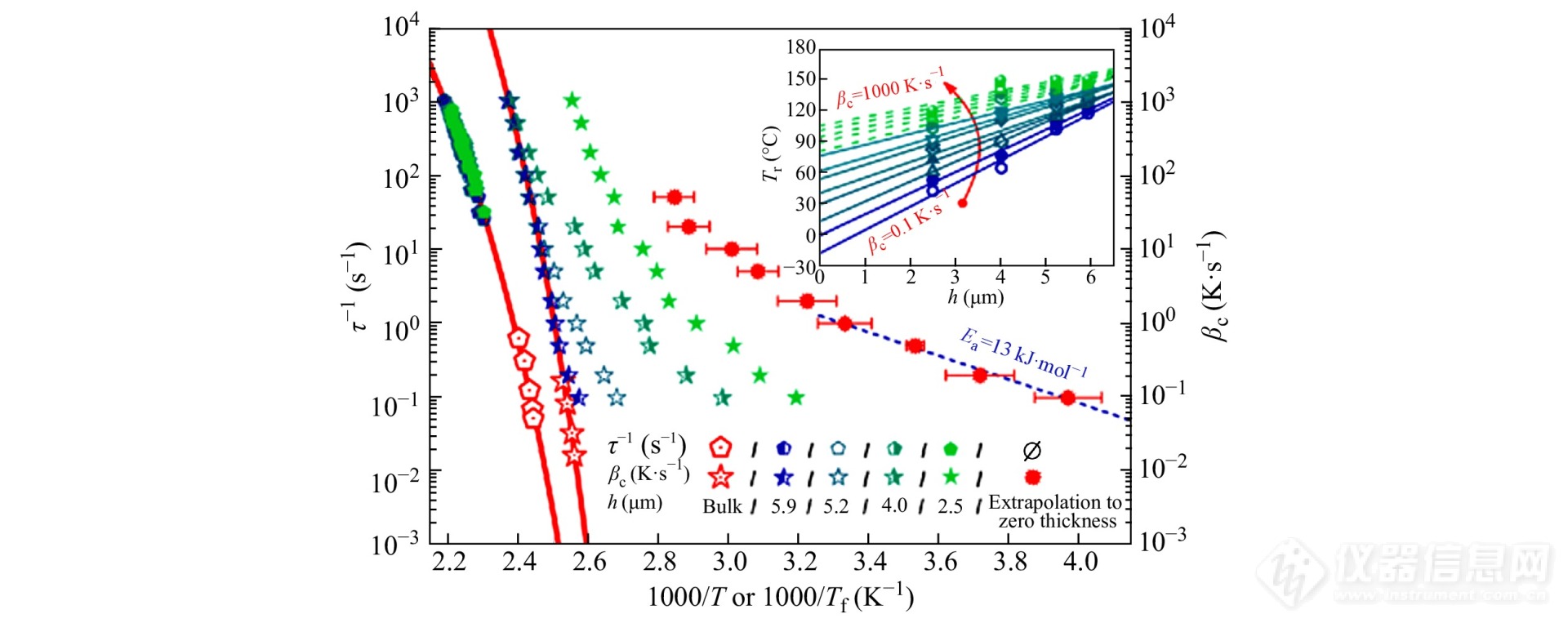
图 26
Figure 26. Reciprocals of the relaxation time (left axis, pentagons) and cooling rate (right axis, stars) as functions of the inverse of temperature and fictive temperature for PtBs samples at different length scales. The solid lines are VFT fits for the relationship between relaxation time (or cooling rate) and fictive temperature. The confinement-length dependence of fictive temperature at different cooling rates is presented in the inset where the dashed and solid lines are linear fits of the length-scale-dependent fictive temperature measured at high and low cooling rates, respectively. (Reprinted with permission from Ref.[87]; Copyright (2014) American Physical Society)
4.3.5 热导率
随着5G时代的来临,电子器件对材料的散热能力要求也越来越高,准确测量材料的热导率对于工业产品质量控制有重要意义. 胡文兵课题组利用FSC技术的优势发展了一种测试微米尺度厚度薄膜材料热导率的新方法[88]. 在薄膜样品上方和参比池上方分别放置一颗铟,然后采用Flash DSC以不同的速率加热样品,通过位于样品上方和参比池上方的铟的熔点之差反映样品上下表面的温差. 根据傅里叶热传导定律可知,样品上下表面的温差与垂直于薄膜表面方向的加热速率成正比,由比例系数可求算样品的热导率. 胡文兵课题组[89,90]采用该方法测量了聚乙烯薄膜样品以及系列尼龙样品的热导率,测得的热导率数值与其采用其他方法测得的文献报道值较为接近,证明了此方法的有效性. 图27为采用该方法表征尼龙610样品热导率得到的熔融曲线. 采用Flash DSC表征材料的热导率具有测试温度和扫描速率范围广、样品量少等优点,该方法还可以表征黏滞液体、取向材料等的导热性能,具有较广阔的应用前景.
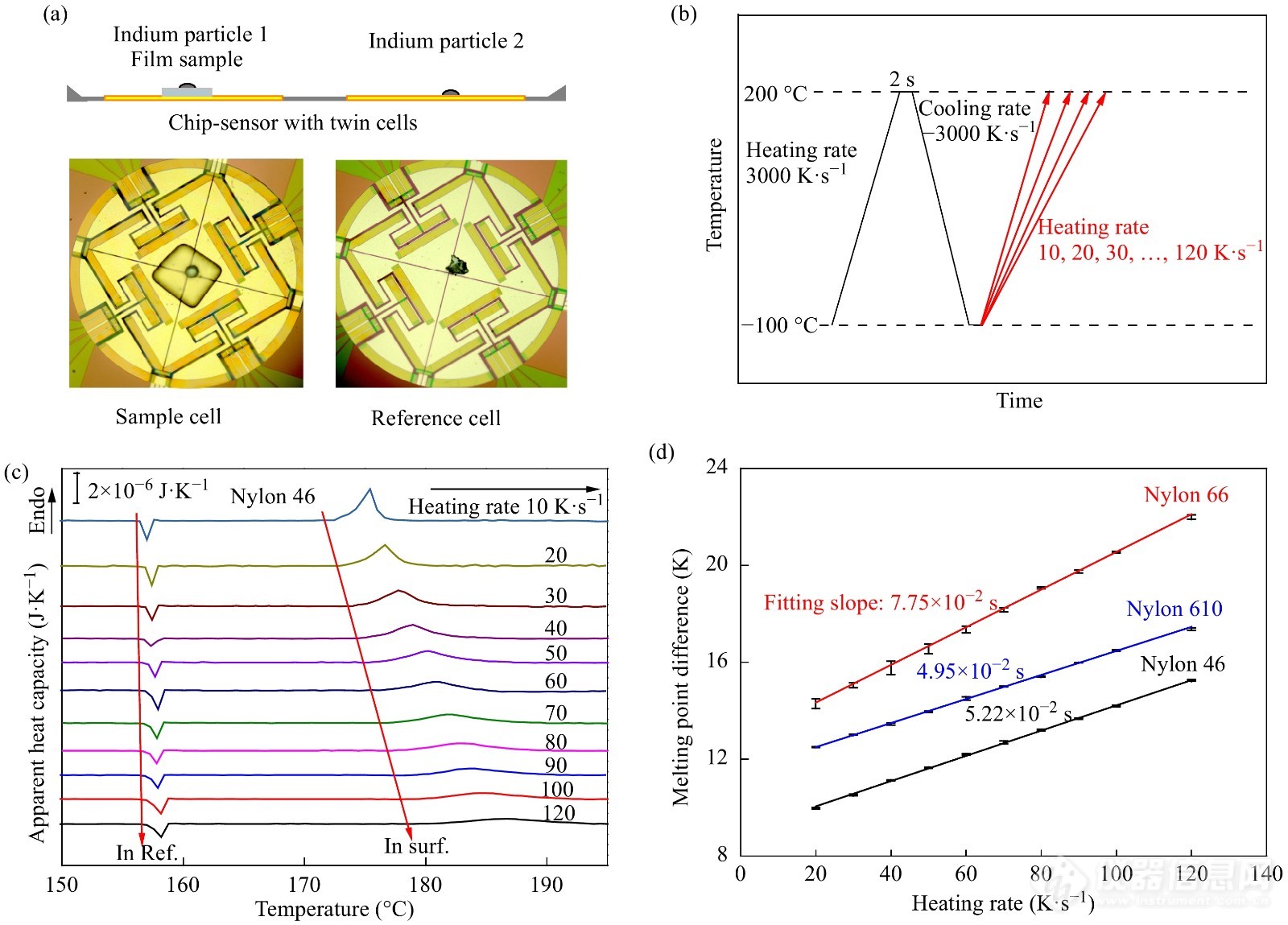
图 27
Figure 27. (a) Top: Illustration of two indium particles separately placed on the top of a regular-shaped sample and on the surface of the reference cell. Bottom: the photographs of the sample cell and the reference cell. (b) Temperature profile for isothermal crystallization and subsequent melting of the samples. (c) Apparent heat capacity curves of Nylon 46 at various heating rates as labeled and the exothermal peak and endothermal peak indicate separately the melting of the indium on the reference cell and on the top of sample Nylon 46. (d) Melting point differences of two indium particles at various heating rates for three Nylon samples (Reprinted with permission from Ref.[90]; Copyright (2014) Elsevier)
5. 总结与展望
本文综述了示差扫描量热法在高分子表征领域的主要进展,旨在帮助大家进一步理解DSC技术的实验原理和方法技巧,探索DSC技术在高分子表征领域的更多应用.
自20世纪60年代以来DSC已经成为了表征材料结构和性能的一种常规研究手段,其在高分子表征领域已经获得了广泛的应用,主要包括在较宽温度范围内测量样品的转变温度和相应的转变焓以及表征玻璃化转变等热容或者潜热发生改变的物理过程,具有操作简便,成本低廉等优点. TMDSC在线性升温速率的基础上叠加了周期性变温速率,保证样品在较长的时间尺度上以一个缓慢的速率升温,同时还能获得一个极快的瞬间温度变化,使得热流信号兼具较高的灵敏度和分辨率,实现了对于微弱转变信号的检测,并能有效区分样品中可逆和不可逆过程的热流信号,甚至准等温过程热容的测量,准确阐明各种转变的本质,为传统DSC的测量结果补充了更多的有效信息. FSC采用氮化硅薄膜传感器取代传统坩埚,将试样量减小到了纳克级别,有效地降低了样品内部的热滞后效应,并实现了106 K·s−1的超快扫描速率. FSC的高扫描速率能抑制高分子在升降温过程中的结构重组,大大推进了对高分子结晶、熔融等相转变过程中非平衡态结构的动力学研究. 同时,FSC将时间窗口缩短到了毫秒级别,能与实际高分子加工过程中的结晶动力学窗口相匹配,有利于加深对高分子加工过程的理解. 此外,FSC将样品体系缩小到微纳米尺度,具有采样损坏小的优点,促进了对纳米空间分辨率的高分子材料内部结构及其性能变化的研究. 总之,DSC已经成为了高分子热分析领域的一项常规表征工具,由其发展出来的FSC技术将其温度扫描速率范围扩展到横跨7个数量级,实现了对从热力学领域的静态热量传递到动力学过程的热量流动速率的一系列表征,有力地推动了高分子基础理论以及加工应用研究的发展.
目前,DSC正朝着更高的扫描速率和更小的样品尺度不断改进和发展,并与其他表征方法更为紧密地连用起来. 如图28所示,分子模拟的时间尺度从纳秒级别自下向上推进,进行理论证明;FSC的时间尺度则自上而下进入到微秒级别进行实验验证,两者的时间窗口在微秒尺度上发生重叠,对应了高分子片晶生长和退火熔融过程的时间尺度. 因此, FSC技术与分子模拟的结合拓宽了其在高分子微观结构表征方面的应用,使人们得以从微观和宏观2个角度研究高分子片晶生长动力学行为. 同时,DSC与其他实验表征手段,如X射线衍射、流变仪、拉曼光谱、偏光显微镜等连用,可以获得在物质的性质发生变化的过程中样品的形貌结构以及机械性能等的变化信息,实现对高分子相转变过程中热力学和动力学现象的多角度深入研究.
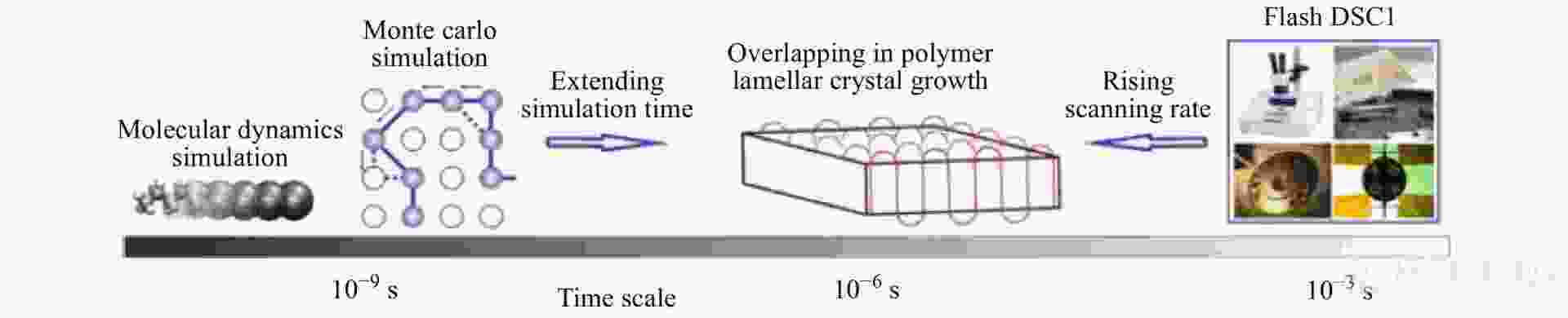
图 28
Figure 28. Illustration of time scales of fast-scan chip-calorimetry measurement and Monte Carlo simulation towards the identical time window of polymer crystallization and melting (Reprinted with permission from Ref.[91]; Copyright (2014) Springer Nature)
参考文献
[1]Rupp R(丽贝卡·鲁普). Water, Gas, Fire and Earth-History of Element Discovery(水气火土—元素发现史话). Beijing(北京): The Commercial Press(商务印书馆), 2008.1−74
[2]ICTAC Nomenclature Committee. Draft-03b. doc 07.03. Recommendations for Names and Definitions in Thermal Analysis and Calorimetry.
[3]ASTM E473-07b, Standard Terminology Relating to Thermal Analysis and Rheology, ASTM International, West Conshohocken, PA, 2007, http://www.astm.org
[4]ASTM E2161-01, Standard Terminology Relating to Performance Validation in Thermal Analysis, ASTM International, West Conshohocken, PA, 2001, http://www.astm.org
[5]Le Chatelier H. Z Phys Chem, 1887, 1: 296
[6]Le Chatelier H. Bull Soc Franc Mineral Cryst, 1887, 10: 204−211
[7]Roberts-Austen W C. Proc Inst Mech Eng, 1899, 1: 35−102
[8]Boersma S L. J Amer Ceram Soc, 1955, 38: 281−284 doi: 10.1111/j.1151-2916.1955.tb14945.x
[9]O’Neill M J. Anal Chem, 1964, 36: 1238−1245 doi: 10.1021/ac60213a020
[10]Watson E S, O’Neill M J, Justin J, Brenner N. Anal Chem, 1964, 36: 1233−1237 doi: 10.1021/ac60213a019
[11]Wunderlich B. Thermal Analysis of Polymeric Materials[M]. Springer: Berlin, 2005. 329−355
[12]Liu Zhenhai(刘振海), Lu Liming(陆立明), Tan Yuanwang(唐远望). A Brief Tutorial on Thermal Analysis(热分析简明教程). Beijing(北京): Science Press(科学出版社), 2012. 83−104
[13]Ding Yanwei(丁延伟). Fundamentals of Thermal Analysis(热分析基础). Hefei(合肥): University of Science and technology of China Press(中国科学技术大学出版社), 2020. 188−231
[14]Lu Liming(陆立明). Basics of Thermal Analysis Application(热分析应用基础). Shanghai(上海): Donghua University Press(东华大学出版社), 2010. 34−43
[15]ASTM E1269-11(2018), Standard Test Method for Determining Specific Heat Capacity by Differential Scanning Calorimetry, ASTM International, West Conshohocken, PA, 2018, http://www.astm.org
[16]Gaur U, Wunderlich B. Advanced thermal analysis aystem (ATHAS) polymer heat capacity data bank. In: Computer Applications in Applied Polymer Science. New York: American Chemical Society, 1982. 355−366
[17]ASTM E1356-08(2014), Standard Test Method for Assignment of the Glass Transition Temperatures by Differential Scanning Calorimetry, ASTM International, West Conshohocken, PA, 2014, http://www.astm.org
[18]Höne G, Hemminger W F, Flammersheim H J. Differential Scanning Calorimetry. Berlin: Springer, 2003. 126−140
[19]Lau S F, Suzuki H, Wunderlich B. J Polymer Sci: Polymer Phys Ed, 1984, 22: 379−405 doi: 10.1002/pol.1984.180220305
[20]Huang M M, Dong X, Wang L L, Zheng L C, Liu G M, Gao X, Li C C, Müller A J, Wang D J. Macromolecules, 2018, 51(3): 1100−1109 doi: 10.1021/acs.macromol.7b01779
[21]Wang Z F, Dong X, Cavallo D, Müller A J, Wang D J. Macromolecules, 2018, 51(15): 6037−6046 doi: 10.1021/acs.macromol.8b01313
[22]Wang Z F, Dong X, Liu G M, Xing Q, Cavallo D, Jiang Q H, Müller A J, Wang D J. Polymer, 2018, 138: 396−406 doi: 10.1016/j.polymer.2018.01.078
[23]Dong Siyuan(董思远), Zhu Ping(朱平), Liu Jiguang(刘继广), Wang Dujing(王笃金), Dong Xia(董侠). Acta Polymerica Sinica(高分子学报), 2019, 50(2): 189−198 doi: 10.11777/j.issn1000-3304.2018.18198
[24]Lu Liming(陆立明). Polymer Bulletin(高分子通报), 2009, (3): 62−74
[25]Corbino O M. Physik Z, 1910, 11: 413−417
[26]Corbino O M. Physik Z, 1911, 12: 292−295
[27]Birge N O, Nagel S R. Phys Rev Lett, 1985, 54: 2674−2677 doi: 10.1103/PhysRevLett.54.2674
[28]Kraftmakher Y A. Z Prikladnoj Mech Tech Fiz, 1962, 5: 176−180
[29]Sullivan P, Seidel G. Ann Acad Sci Fennicae A VI, 1966, 210: 58−62
[30]Gobrecht H, Hamann K, Willers G. J Phys E: Sci Instrum, 1971, 4: 21−23 doi: 10.1088/0022-3735/4/1/004
[31]Gill P S, Sauerbrunn S R, Reading M. J Therm Anal, 1993, 40: 931−939 doi: 10.1007/bf02546852
[32]Reading M, Elliott D, Hill V L. J Thermal Anal, 1993, 40: 949 doi: 10.1007/BF02546854
[33]Reading M, Luget A, Wilson R. Thermochim Acta, 1994, 238: 295−307 doi: 10.1016/S0040-6031(94)85215-4
[34]Reading M, Hourston D J. Modulated Temperature Differential Scanning Calorimetry: Theoretical and Practical Applications in Polymer Characterization. Berlin: Springer, 2006. 1−80
[35]Hu W B, Wunderlich B. J Therm Anal Calorim, 2001, 66: 677−697 doi: 10.1023/A:1013106118660
[36]Wunderlich B. Prog Polym Sci, 2003, 28: 383−450 doi: 10.1016/S0079-6700(02)00085-0
[37]Schick C, Wurm A, Mohammed A. Thermochim Acta, 2003, 396: 119−132 doi: 10.1016/S0040-6031(02)00526-9
[38]Schick C. Anal Bioanal Chem, 2009, 395: 1589−1611 doi: 10.1007/s00216-009-3169-y
[39]Hu W B, Albrecht T, Strobl G. Macromolecules, 1999, 32: 7548−7554 doi: 10.1021/ma9908649
[40]Jiang X M, Li Z L, Wang J, Gao H H, Zhou D S, Tang Y W, Hu W B. Thermochim Acta, 2015, 603: 79−84 doi: 10.1016/j.tca.2014.04.002
[41]Gaur U, Wunderlich B. J Phys Chem Ref Data, 1981, 10: 1051−1064 doi: 10.1063/1.555650
[42]Boller A, Schick C, Wunderlich B. Thermochim Acta, 1995, 266: 97−111 doi: 10.1016/0040-6031(95)02552-9
[43]Pyda M, Wunderlich B. Macromolecules, 2005, 38(25): 10472−10479 doi: 10.1021/ma051611k
[44]Schick C, Mathot V B F. Fast Scanning Calorimetry[M]. Springer: Switzerland, 2016. V−VII
[45]Li Zhaolei(李照磊), Zhou Dongshan(周东山), Hu Wenbing(胡文兵). Acta Polymerica Sinica(高分子学报), 2016, (9): 1179−1195 doi: 10.11777/j.issn1000-3304.2016.16058
[46]Denlinger D W, Abarra E N, Allen K, Rooney P W, Messer M T, Watson S K, Hellman F. Rev Sci Instrum, 1994, 65(4): 946−958 doi: 10.1063/1.1144925
[47]Allen L H, Ramanath G, Lai S L, Ma Z, Lee S, Allman D D J, Fuchs K P. Appl Phys Lett, 1994, 64(4): 417−419 doi: 10.1063/1.111116
[48]Efremov M Yu, Olson E A, Zhang M, Schiettekatte F, Zhang Z S, Allen L H. Rev Sci Instrum, 2004, 75(1): 179−191 doi: 10.1063/1.1633000
[49]Adamovsky S, Minakov A A, Schick C. Thermochim Acta, 2003, 403(1): 55−63 doi: 10.1016/S0040-6031(03)00182-5
[50]Adamovsky S, Schick C. Thermochim Acta, 2004, 415(1-2): 1−7 doi: 10.1016/j.tca.2003.07.015
[51]Yu J, Tang Z A, Zhang F T, Wei G F, Wang L D. Chin Phys Lett, 2005, 22(9): 2429−2432 doi: 10.1088/0256-307X/22/9/080
[52]Jiang J, Wei L, Zhou D. Integration of Fast Scanning Calorimetry (FSC) with microstructural analysis techniques. In: Schick C, Mathot V B F, ed. Fast Scanning Calorimetry, Switzerland: Springer, 2016. 361−379
[53]Chen M Z, Du M T, Jiang J, Li D W, Jiang W, Zhuravlev E, Zhou D S, Schick C, Xue G. Thermochim Acta, 2011, 526(1-2): 58−64 doi: 10.1016/j.tca.2011.08.020
[54]Jiang J, Zhuravlev E, Huang Z, Wei L, Xu Q, Shan M, Xue G, Zhou D, Schick C, Jiang W. Soft Matter, 2013, 9(5): 1488−1491 doi: 10.1039/C2SM27012A
[55]Wei L, Jiang J, Shan M, Chen W, Deng Y, Xue G, Zhou D. Rev Sci Instrum, 2014, 85(7): 074901−074907 doi: 10.1063/1.4889882
[56]van Herwaardena S. Procedia Eng, 2010, 5: 464−467 doi: 10.1016/j.proeng.2010.09.147
[57]Schick C, Mathot V B F. Material Characterization by Fast Scanning Calorimetry: Practice and Applications. In Fast Scanning Calorimetry. Switzerland: Springer, 2016. 3−299
[58]Wang J, Li Z L, Perez R A, Müller A J, Zhang B Y, Grayson S M, Hu W B. Polymer, 2015, 63: 34−40 doi: 10.1016/j.polymer.2015.02.039
[59]Zhuravlev E, Schmelzer J W P, Wunderlich B, Schick C. Polymer, 2011, 52: 1983−1997 doi: 10.1016/j.polymer.2011.03.013
[60]Kalapat D, Tang Q Y, Zhang X H, Hu W B. J Therm Anal Calorim, 2017, 128: 1859−1866 doi: 10.1007/s10973-017-6095-9
[61]Santis F D, Adamovsky S, Titomanlio G, Schick C. Macromolecules, 2006, 39: 2562−2567 doi: 10.1021/ma052525n
[62]Santis F D, Adamovsky S, Titomanlio G, Schick C. Macromolecules, 2007, 40: 9026−9031 doi: 10.1021/ma071491b
[63]Stolte I, Androsch R, Di Lorenzo M L, Schick C. J Phys Chem B, 2013, 117(48): 15196−15203 doi: 10.1021/jp4093404
[64]Shick C, Androsch R. New insights into polymer crystallizaiton by fast scanning chip calorimetry. In: Fast Scanning Calorimetry. Switzerland: Springer, 2016. 463−537
[65]He Yucheng(何裕成), Xie Kefeng(谢科锋), Wang Youhao(王优浩), Zhou Dongshan(周东山), Hu Wenbing(胡文兵). Acta Physico-Chimica Sinica(物理化学学报), 2020, 36(6): 1905081−1905092 doi: 10.3866/PKU.WHXB201905081
[66]Zhuravlev E, Schick C. Thermochim Acta, 2010, 505(1-2): 1−13 doi: 10.1016/j.tca.2010.03.019
[67]Mollova A, Androsch R, Mileva D, Gahleitner M, Funari S S. Eur Polym J, 2013, 49(5): 1057−1065 doi: 10.1016/j.eurpolymj.2013.01.015
[68]Iervolino E, van Herwaarden A W, van Herwaarden F G, van de Kerkhof E, van Grinsven P P W, Leenaers A C H I, Mathot V B F, Sarro P M. Thermochim Acta, 2011, 522(1-2): 53−59 doi: 10.1016/j.tca.2011.01.023
[69]Cebe P, Partlow B P, Kaplan D L, Wurm A, Zhuravlev E, Schick C. Thermochim Acta, 2015, 615: 8−14 doi: 10.1016/j.tca.2015.07.009
[70]Wang T, Li X H, Luo R Q, He Y C, Maeda S, Shen Q D, Hu W B. Thermochim Acta, 2020, 690: 178667−178672 doi: 10.1016/j.tca.2020.178667
[71]Hu Wenbing(胡文兵). Principles of Polymer Crystallization(高分子结晶学原理). Beijing(北京): Chemical Industry Press(化学工业出版社), 2013. 114−163
[72]Hu Wenbing(胡文兵). Introduction to Polymer Physics(高分子物理导论). Beijing(北京): Science Press(科学出版社), 2011. 146−173
[73]He Y C, Luo R Q, Li Z L, Lv R H, Zhou D S, Lim S, Ren X N, Gao H X, Hu W B. Macromol Chem Phys, 2018, 219: 1700385−1700390 doi: 10.1002/macp.201700385
[74]Hu W B. Phys Rep, 2018, 747: 1−50 doi: 10.1016/j.physrep.2018.04.004
[75]Li X H, He Y C, Dong X, Ren X N, Gao H X, Hu W B. Polymer, 2020, 189: 122165−122173 doi: 10.1016/j.polymer.2020.122165
[76]Toda A, Mikosaka M, Yamada K. Polymer, 2002, 43: 1667−1679 doi: 10.1016/S0032-3861(01)00733-9
[77]Toda A, Kojima I, Hikosaka M. Macromolecules, 2008, 41: 120−127 doi: 10.1021/ma702162m
[78]Gao H H, Wang J, Schick C, Toda A, Zhou D S, Hu W B. Polymer, 2014, 55(16): 4307−4312 doi: 10.1016/j.polymer.2014.06.048
[79]Monnier X, Napolitano S, Cangialosi D. Nat Commun, 2020, 11: 4354−4360 doi: 10.1038/s41467-020-18216-y
[80]Huang Z J, Jiang J, Xue G, Zhou D S. Chinese J Polym Sci, 2019, 37: 94−100 doi: 10.1007/s10118-019-2177-4
[81]Luo S C, Wei L, Jiang, J, Sha Y, Xue G, Wang X L, Zhou D S. J Polym Sci, Part B: Polym Phys, 2017, 55: 1357−1364 doi: 10.1002/polb.24378
[82]Luo S C, Kui X, Xing E R, Wang X L, Xue G, Schick C, Hu W B, Zhuravlev E, Zhou D S. Macromolecules, 2018, 51(14): 5209−5218 doi: 10.1021/acs.macromol.8b00692
[83]Luo S C, Wang T Y, Ocheje M U, Zhang S, Xu J, Qian Z Y, Gu X D, Xue G, Rondeau-Gagné S, Jiang J, Hu W B, Zhuravlev E, Zhou D S. Macromolecules, 2020, 53(11): 4480−4489 doi: 10.1021/acs.macromol.9b02738
[84]Jiang J, Zhuravlev E, Hu W B, Schick C, Zhou D S. Chinese J Polym Sci, 2017, 35(8): 1009−1019 doi: 10.1007/s10118-017-1942-5
[85]Lv R H, He Y C, Wang J P, Wang J, Hu J, Zhang J M, Hu W B. Polymer, 2019, 174: 123−129 doi: 10.1016/j.polymer.2019.04.061
[86]Schawe Jürgen E K, Löffler Jörg F. Nat Commun, 2019, 10(1): 1337−1346 doi: 10.1038/s41467-018-07930-3
[87]Monnier X, Cangialosi D. Phys Rev Lett, 2018, 121: 137801−137806 doi: 10.1103/PhysRevLett.121.137801
[88]Zhang Jianjun(张建军). Acta Physico-Chimica Sinica(物理化学学报), 2020, 36(6): 1907048−1907049 doi: 10.3866/PKU.WHXB201907048
[89]He Y C, Li X H, Ge L, Qian Q Y, Hu W B. Thermochim Acta, 2019, 677: 21−25 doi: 10.1016/j.tca.2019.01.003
[90]Xie K F, He Y C, Cai J, Hu W B. Thermochim Acta, 2020, 683: 178445−178449 doi: 10.1016/j.tca.2019.178445
[91]Jiang X M, Li Z L, Gao H H. Combining fast-scan chip calorimetry with molecular simulation to investigate polymer crystal melting. In: Schick C, Mathot V B F, ed. Fast Scanning Calorimetry. Springer: Switzerland, 2016. 379−403
来源于:仪器信息网

相关阅读
TA仪器与华南理工大学材料科学与工程学院新建联合实验室
Sunshine



“聚”先锋 | 用热分析和流变学优化3D打印
Sunshine

2024德国设计奖揭晓 3款仪器产品脱颖而出
Sunshine

仪器优选·差示扫描量热仪
更多




近期会议
更多



推荐专题

热门评论
最新资讯
新闻专题
更多推荐
