
技术漫谈|超高分辨率显微成像技术在神经科学中的应用(一)


导读:今年春,Werner等科学家在美国化学学会会刊(ACS)上最新发表了一篇综述,比较详实系统介绍了超高分辨率显微技术在神经科学上的最新应用进展。我们在此文基础上进行了编译整理。
荧光显微成像技术对人们理解神经科学起了非常关键的作用。而最近一些年出现的各种超分辨显微成像技术和专门的荧光探针能够以超过以往普通光学显微镜的分辨率直接观察神经元亚细胞结构和蛋白质排列。并以直观可视方式揭示了神经细胞骨架组成、分布、运动和膜蛋白信号传导、突触下结构和功能,以及神经元−胶质细胞相互作用。同时超高分辨显微成像技术(Super Resolution,SR,下文中出现SR均指超高分辨率显微成像技术)对于许多自身免疫和神经退行性疾病模型中的分子靶点研究也提供了全新的强大工具。今年春,Werner等科学家在美国化学学会会刊(ACS)上最新发表了一篇综述,比较详实系统介绍了超高分辨率显微技术在神经科学上的最新应用进展。我们在此文基础上进行了编译整理。因文章较长,我们将分三期陆续介绍。本期介绍第一部分。
1. 背景介绍
成像技术是推动生命科学几乎所有学科基础研究的核心平台。在神经科学领域,近几十年来,共聚焦显微镜技术已成为分析神经组织的标准荧光成像技术。激光扫描共聚焦显微镜对固定的神经元样本进行观察,在扫描水平上提供了三维和多色图像并使单个细胞达到树突结构的分辨率。作为补充,电子显微镜(EM)用于获取神经元和亚区室超微结构的信息,并用于大脑的连通性分析。EM非常适合于神经元突触和囊泡、细胞器和膜构象的结构分析。
然而,由于靶向特异性标记方法的局限性,基于EM的复杂样品中蛋白质和特定电子密度特征的识别受到限制。为了进一步理解神经元功能,包括双光子显微镜在内的几种活体视频显微镜应用的发展使神经元细胞培养的活细胞成像、器官型切片培养和动物模型的活体成像成为可能。同时,新的荧光染料、功能探针和荧光蛋白以及光遗传学方法和光驱动(如笼状化合物)不仅可以表征神经元,还可以操纵神经元及其从单分子水平到整个神经系统的相互作用。
然而,荧光显微图像中可见细节的水平,即图像分辨率,仍然受到衍射极限的限制。一个多世纪以来,由λ/2NA定义的阿贝衍射极限(λ为波长,NA为显微镜物镜的数值孔径)决定了光学显微镜的分辨率极限,限制了两个位置小于200纳米的细节分辨。在过去的二十年中,超分辨显微镜(SRM)已经发展成为一种非常有效的亚细胞水平荧光成像和分辨细胞器结构的研究手段。SRM现在可以提供远低于常规光学显微镜衍射极限的空间分辨率,从而能够深入了解神经元细胞和组织中蛋白质的空间结构和相互作用。
本文综述了超分辨显微镜和荧光标记方法及其在神经科学中的成功应用。我们将首先详细介绍各种SRM方法的基本原理、新的功能型荧光探针和标记技术。接着,我们将回顾SRM如何有助于我们理解神经元亚细胞结构和功能以及神经元−胶质细胞相互作用。此外,我们将概述超分辨率成像方法如何帮助研究自身免疫和神经退行性疾病的病理生理学。最后,我们将介绍这些新的成像方法是如何应用于神经精神疾病相关的人类样本的分析。由于该领域持续快速发展,我们最多只能代表一份中期报告。进一步的创新和新的显微镜方法的发展将使人们对神经系统功能有更详细的了解。
2. 神经科学中的超分辨率成像方法
2.1. 光学衍射极限及其对神经科学的影响
人类大脑包含超过800亿个神经元,每个神经元由数千个突触连接。因此,它构成了复杂神经元网络。这些网络的主要组成部分,例如突触神经末梢,显示的空间维度接近于光学衍射极限分辨率∼200 nm。释放递质的突触活性区(突触前细胞基质的特化区)的直径通常约为300±150 nm。突触小泡作为递质运输和释放的关键元件,其尺寸平均小10倍,直径为40−50nm。这些递质被释放到宽度为20-50nm的突触间隙中−再结合突触后受体。由于衍射极限的尺寸限制,胞吐机制和跨突触信号在传统的光学显微镜下基本上是无法观测到的,因此需要用提高10倍分辨率的方法进一步研究。(图1)。

图1. 兴奋性突触结构组成。左图为兴奋性突触的油画示意图,右图为左图的灰度图像,其中浅紫色圆圈为衍射极限光斑;玫红色圆圈为兴奋性突触囊泡,约40-50nm;绿色为突触后膜AMPA受体,尺寸小于10nm;黄色部分为突触间隙,约20-30nm。
此外,大量参与突触信号传导的不同的分子,位于极小的突触内,造成很高的分子分布密度,这对微观研究具有挑战性。例如,对于较小的突触,兴奋性突触可以包含数百个小泡,对于大型苔藓纤维束突触,可以包含数千个小泡,每个小泡包含多达1万到10万个递质分子。在这些囊泡中,约有10±5个与释放部位对接,释放的递质平均与0−20 个NMDA受体和0−200个AMPA受体结合,而这些突触后受体又被320±130个突触后PSD-95密度蛋白分子环绕。由于加速电子的波长要短得多,因此EM是唯一能够解析突触纳米级结构的方法。然而,虽然传统的EM产生的电子密度图像具有极好的超微结构分辨率,但需要进行固定和靶向特异性标记的制样方法在很大程度上限制了蛋白质识别和神经元追踪。荧光显微镜可以很容易地对蛋白质进行选择性标记,但是受制于可见光的衍射(400−700 nm)使生成的图像无法实现对纳米结构的分析。
2.2.绕开光学衍射极限的光学显微镜方法
20世纪后期,人们开发了新的策略,通过利用物理或化学手段来区分不同荧光团的发射或减少同一时间荧光分子的数量,以尽量绕过衍射极限。减少荧光团的点扩散函数(PSF)的重叠可以通过生成光图案在集合级别以确定性方式进行,或者通过减少同一时间荧光团的数量在单分子水平上以随机方式进行。在下文中,我们将从确定性集合方法开始介绍,该方法将激光扫描共聚焦显微镜(CLSM)的有效空间分辨率推到理论极限。
2.2.1. 确定性集合超高分辨率成像方法(Deterministic Ensemble SR-Imaging Methods)
CLSM用针孔探测器阵列替换单点探测器,空间分辨率可以提高√2倍。CLSM测量每个扫描位置探测器每个点的荧光信号。在应用适当的算法后,生成分辨率提升的图像。这些所谓的像素重分配方法包括图像扫描显微镜(ISM)、重扫描共聚焦(RSC)、光学光子重分配(OPRA)、AiryScan和即时结构照明显微镜(iSIM)。对于信号检测,使用了诸如CCD相机、光电倍增管阵列、单光子雪崩二极管阵列和六角光纤束等探测器阵列。
结构照明显微镜(SIM)在光路中插入光栅,产生与样品干涉的相干光束,生成横向和轴向方向不同的新照明图案。然后可以使用傅里叶变换提取这种新照明图案的信息,从而在所有三维空间中实现空间频率分解和分辨率倍增。SIM对样品制备的要求最低,并且可使用所有常规荧光探针,这些探针具有最低的光稳定性,并且可以很容易地扩展到多色成像。
然而,当记录三维或长时间成像时,强烈建议使用光稳定性更高的荧光团。此外,SIM使用更低的激发强度,因此是活细胞SR实验的理想选择。为了获得更高的分辨率,引入了通过图案化饱和或荧光激发或图案化耗损光开关染料的非线性SIM(NL-SIM)。然而对染料开关特性的苛刻要求限制了NL-SIM在常规生命科学实验中的适用性。非线性SIM单位时间内还需要采集更多的图像,因此实际上仅限于2D成像。
另一方面,掠入射(GI)-SIM显示了高达每秒266帧的快速超分辨率成像以及100nm分辨率,揭示前所未有的细胞器动力学细节。结构照明的局限性在于其对波长的普遍依赖性、与其他SR成像技术相比的低分辨率以及对系统稳定校准的需要。最后,后处理需要进行先验质量检查以避免伪影,例如由于高背景信号或不充分标记产生的低对比度图像导致的人工蜂窝图案。
通过受激发射耗损(STED)显微镜进行超分辨率成像是一种实现更高空间分辨率的成像方法。这里,高斯分布的激发激光束被中空的甜甜圈样的耗损激光束覆盖,使扫描点外围的荧光团返回基态,这导致纳米级焦点区的直径与耗损光束的强度成反比,耗损光束的强度直接转换为STED显微镜的分辨能力:
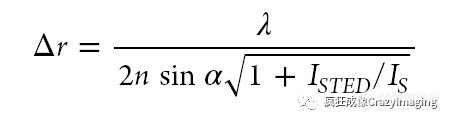
上图公式中λ为波长,n为折射率,α为物镜的收集角,ISTED为STED光束的照射强度,IS为饱和强度。
因此,可以通过改变损耗激光强度来调整分辨率,可定制设计分辨率达30−80nm 的显微镜。STED显微成像可通过连续或脉冲激光激发、门控检测。带有脉冲激光的STED显微镜会降低激发能量,从而减少实时成像中的光毒性效应。STED显微镜中的时间门控检测可以去除荧光团光子到达时间前的空间信息,并且可以在较低的平均功率下工作。商品化STED能提供用户友好的高分辨率成像,无需进一步的数据后处理。活体成像,例如活体树突棘动态成像已经很成熟,但快速动态成像仅限于小帧尺寸,因为它仍然是点扫描方法,高激光强度可能会导致光损伤。STED通过应用自适应照明方式Dymin和rescue技术,可以明显减少光损伤。
在Dymin STED中,在共聚焦模式下扫描时确定最低可能的STED光束强度。根据样品的标记密度,这将使STED光束强度降低20到100倍。Rescue STED同样通过减少STED激光开放的区域,从而比普通STED减少光漂白接近8倍。STED的另一个限制是对荧光团光稳定性的依赖,因为在高激光强度下会发生明显的光漂白。这影响了动力学的研究和三维图像的获取。值得注意的是,最近通过使用荧光团标记的寡核苷酸(瞬时结合到连接靶蛋白结合探针的互补寡核苷酸)或非结合荧光团来进行细胞STED成像,从而绕过了STED光漂白问题。这两种方法中,基于DNA互补标记的STED成像和超分辨率阴影成像SUSHI分别通过荧光团标记的寡核苷酸和高浓度的非结合和自由扩散的荧光团不断交换来防止光漂白。SUSHI的方法已经成功地用于活体脑片中细胞外间隙和神经肽的结构解析及其动力学的STED成像。如果使用具有毫秒或更长寿命的两种稳定状态的可逆切换荧光团来代替标准荧光团,则STED强度可以显著降低。可逆饱和切换光学线性荧光转换方法(RESOLFT)已通过可逆可切换荧光蛋白(reFPs)实现,并成功应用于活体海马脑片树突棘的超分辨率成像。
2.2.2. 随机单分子SR成像方法(Stochastic Single-Molecule SR-Imaging Methods)
上述的确定性方法是通过改变激发模式或相位掩膜来暂时控制荧光发射达到超分辨成像,而基于单分子的定位SR显微镜则是随机地在时间上分离单个荧光团的发射。
单分子定位显微镜(SMLM)基于单个荧光团的随机激活,使用配备高灵敏相机(EMCCD或sCMOS)的宽场荧光显微镜进行单分子检测,以及精确的位置测定。通过将理想PSF与实际测量的光子分布拟合来进行分子定位。只要信号来自单个发射区,且单个发射区之间的距离大于显微镜能分辨的最小距离,则通过收集更多光子和最小化噪声,定位的标准误差可以任意小。激活和定位过程重复多次,所有定位最终用于重建超分辨率图像。
为了确保在成像的任何时候,只有稀疏的小荧光团以其活性荧光形式存在(开启状态),使用了光开关、光转换、光激活或自发闪烁的荧光团。由于定位精度和最终图像分辨率取决于每次检测到的光子数量,通常采用明亮且稳定的荧光团与>1 kW/cm2的辐照强度相结合的方式。根据所使用的荧光团不同,SMLM可达到10−50 nm横向分辨率。光激活荧光蛋白(FPs),自2006年以来已用于光激活定位显微镜(PALM),例如在405 nm的激光照射下可从关闭状态不可逆地转换为打开状态的PA-GFP和PA-mCherry; 以及可通过适当波长的激光照射从一种波长状态不可逆地转移到另一种波长状态的光转换FPs,例如MEO。此外,还成功地应用了诸如Dronpa之类的光开关FPs,其在不同激发波长的激光照射下可在非荧光和荧光状态之间可逆地切换。对于活细胞应用,使用荧光蛋白的PALM是首选方法。因为在理想情况下,每个感兴趣的蛋白质都可以用荧光蛋白进行计量标记。然而,荧光蛋白比有机染料表现出更低的光稳定性和光子计数,从而降低了定位精度,并且通常需要更长的采集时间。此外,对于PALM成像而言,融合蛋白通常会过度表达,这可能会导致不真实图像,而用转基因变体替代显示野生型表达和功能的自身蛋白仍然具有挑战性。
对于细胞内源性蛋白质的标记,通常使用有机染料的免疫标记。SMLM适用的有机染料必须是光开关、光激活或自发闪烁的,以实现单个染料发射的时间分离,但化学计量标记要困难得多。有机染料通常表现出较高的光子计数和光稳定性,从而使定位精度达到5−10nm。花菁染料Cy5和Alexa Fluor 647可以在荧光开启状态(其典型寿命为10 ms)和非荧光关闭状态(寿命为几秒,利用光开关缓冲液,缓冲液包括PBS,10−100mM硫醇,如ß-巯基乙缅(MEA),酶促氧清除剂,可以有/没有激活染料)之间可逆切换,为随机光学重建显微镜(STORM)和直接型STORM(dSTORM)的发展铺平了道路。近年来,应用于(d)STORM的染料已大大扩展,除了菁染料外,还包括罗丹明和恶嗪染料。有趣的是,最近的研究表明,即使是多个标记的抗体在光开关缓冲液中也呈现出类似于单发射的表现,因此适用于dSTORM实验。
光活化染料的作用与光活化荧光蛋白相似。也就是说,它们在被光照射或自发激活之前处于非荧光状态。罗丹明衍生物PA-JF549和PA-JF646以及桥环菁染料Cy5B是已成功用于SMLM的光活化染料。此外,在没有光开关缓冲液的水溶液中,硅罗丹明HMSiR等自发闪烁染料也能应用于SMLM。最近,通过图案化照明方式实现更高的定位精度,单个荧光发射区的定位得到了改进。定位精度取决于信号的大小和强度,可以通过测量的PSF标准偏差的平方除以收集的光子数来估计。然而,包括拟合性能、标记密度、标记误差和显微镜漂移在内的其它参数决定了高定位精度是否可以转化为低于10 nm的空间分辨率。此外,到目前为止,因为SMLM方法成像需要昂贵的仪器和成像者具备广泛的专业知识,这在一定程度上阻碍了其广泛应用。
2.2.3. SMLM-点累计纳米成像技术(PAINT,Point Accumulation for Imaging Nanoscale Topography)
第一代SMLM技术依赖于荧光团的光开关和光激活,其分辨率需要有效地利用荧光团发出的光子数,而PAINT(point accumulation for imaging nanoscale topography)方法使用活的,与目标区域结构短瞬结合的染料。在成像过程中,被漂白的荧光团可以被成像介质中充足的新鲜荧光团不断置换替补。
由于游离染料在采集单个图像帧期间在多个像素上快速扩散,因此它们仅显示为模糊背景且不能准确定位,而结合染料显示为PSF且能准确定位。因此PAINT的第一种方法是将荧光染料(如尼罗红)与细胞膜进行非特异性结合,然后进行光漂白和新的结合。此外,基于蛋白质片段的探针被用于单分子定位标记。在最近的一个研究中,将这种方法与传统的基于phalloidin的肌动蛋白标记方法进行了比较。通过引入通用PAINT(uPAINT)使Ni-Tris-NTA与转基因蛋白质上表达的His-Tags更特异结合,并可用于突触间隙成像。uPAINT也可以应用于其它标记方法,如免疫标记(内源性蛋白抗体、纳米抗体如绿色荧光蛋白)或受体配体结合。
为了提高PAINT的适用性和特异性,引入DNA-PAINT方法。它使用长度小于10个核苷酸的短的可控的寡核苷酸链(成像链)瞬时标记其靶结合互补寡核苷酸链(对接链)。成像链与对接链的瞬时结合产生明显的闪烁。因此,荧光团开-关状态之间的切换与其光物理性质不直接关联。DNA-PAINT首先在DNA折纸(DNA-origami)上得到验证。DNA折纸是一种自组装的DNA结构(具有已知的大小),通过侧链和荧光团进行结合,并通过宽场显微镜观察。
总的来说,DNA-PAINT是一种易于实现的SR成像标记方法,无需特定光物理特性的荧光团。因为探针可以在一轮结合后,从成像介质中置换补充荧光团,从而避免了光漂白。DNA-PAINT的缺点是图像获取时间长,这是由成像链与对接链的结合和解离速率决定的,以及荧光成像链的纳摩尔浓度引起的背景信号。尽管通过使用优化的DNA序列和缓冲条件,以及使用串联的周期性DNA结构域或通过短肽的卷曲螺旋相互作用(称为“Peptide-PAINT”),可以加快采集速度,但还是要利用全内反射荧光(TIRF)(仅限于对靠近盖玻片结构进行成像的特点),才能更好地减少成像链的背景信号。
另一方面,基于DNA的探针提供了序列成像复用的明显优势,如Exchange PAINT中所述,已成功用于小鼠视网膜切片中多个结构的成像(图2)。Exchange PAINT的概念也被推广到dSTORM、STED、SIM和更传统的衍射限制的宽场和共聚焦荧光显微镜。最近,通过一种称为PRISM(probe-based imaging for sequential multiplexing)的基于DNA-PAINT的成像方法,实现了高达10个神经元蛋白质的分辨率约为20nm的多通道成像。该方法使用了低亲和力成像探针,该探针与突触、肌动蛋白和微管一抗上的对接链结合。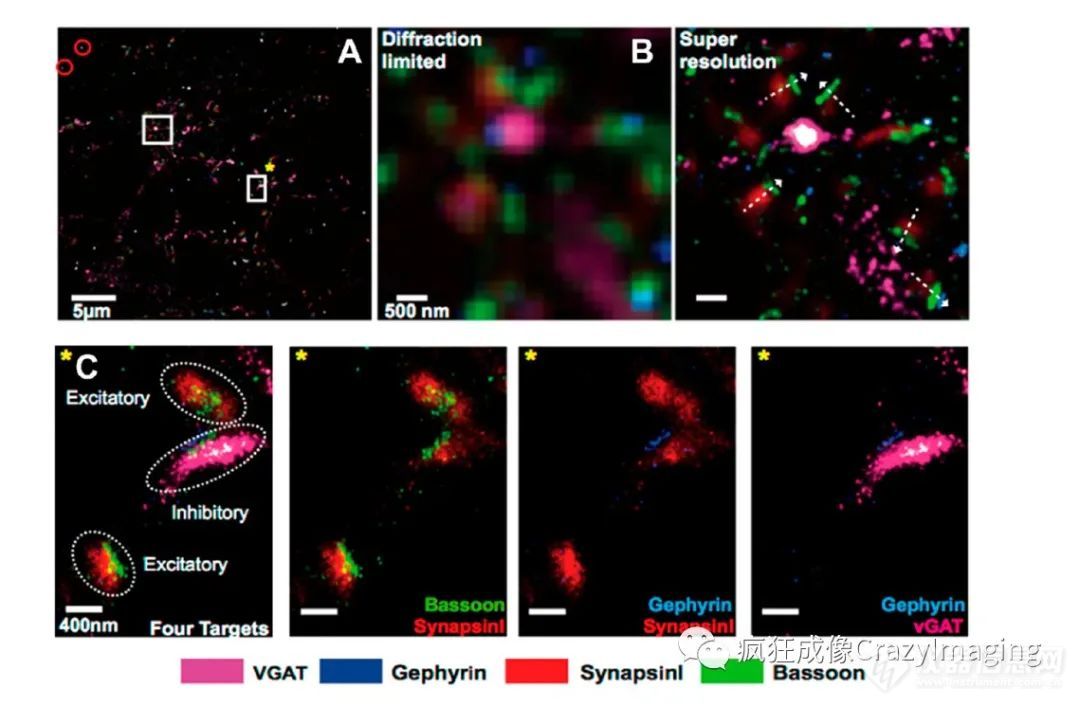
图2 原代神经元中多个神经元靶点的多标Exchange-PAINT成像。(A)DNA-PAINT顺序成像的四种突触蛋白的超分辨图像:圆圈表示漂移校正的基准点;(B)为(A)中不带*的感兴趣区域的高放大倍率图和超分辨图像。(C)为(A)中带*的感兴趣区域的超分辨结果及单通道图像。
2.2.4. 定量SMLM
如果每个目标分子都可以单独标记和定位的话,与所有其他超分辨率成像技术相比,SMLM还可以提供有关分子分布和分子绝对数的单分子信息。然而,内源性蛋白质的定量免疫标记仍然是一个挑战,并且多标记抗体的不同定位数目也会使数据解释复杂化。另一方面,达到内源性表达水平比较困难,另外FPs蛋白成熟缓慢也同样会令定量化困难。
然而,可以通过设计专门的对照实验估计拷贝数,并提取出有关生物目标结构分子的真实信息。借助合适的算法,SMLM可以提供有关拷贝数、聚类、共定位和复杂化学计量的数据,用于定量模型的生成和模拟。此外,还可以通过将突触结构信息与其功能关联来实现量化,例如膜片钳神经元的生物细胞素标记。例如,通过对链霉亲和素标记后膜片钳神经元进行STORM成像,结合CB1受体的免疫标记,然后在GABA能的海马轴突终端内定量,研究了内源性大麻素信号。
本研究发现,与树突投射型中间神经元相比,胞周投射型中间神经元具有更高的CB1受体密度和更复杂的活动区。通过免疫标记和dSTORM研究了黑腹果蝇神经肌肉连接处内源性Bruchpilot(Brp)分子的数量。利用抗体滴定实验,确定了野生型神经肌肉连接处活性区细胞基质中Brp蛋白的数量为137个,其中四分之三以约15个七聚体簇状排列结合从相同组织样本记录的电生理数据,研究Brp如何组织控制活动区功能。利用DNA纳米结构作为校准,每个活性区Brp蛋白的数量估计通过定量DNA-PAINT(qPAINT)实验证实。此外,定量dSTORM实验表明,每个活性区Brp蛋白的数量和分布受突触标记蛋白-1的影响,这说明突触活性区递质释放的复杂性。
在最近的一项研究中,使用Alexa Fluor 532和Alexa Fluor 647免疫标记的双色dSTORM已用于小鼠小脑平行纤维活性区中代谢型谷氨酸受体4(mGluR4)的定量研究(图3)。该研究还使用抗体滴定实验估计每个活性区平均包含约35个mGluR4分子,并排列在小纳米结构中。此外,mGluR4通常在munc-18-1和CaV2.1通道附近被发现,这支持了mGluR4与这些蛋白质相互作用以调节突触传递的观点。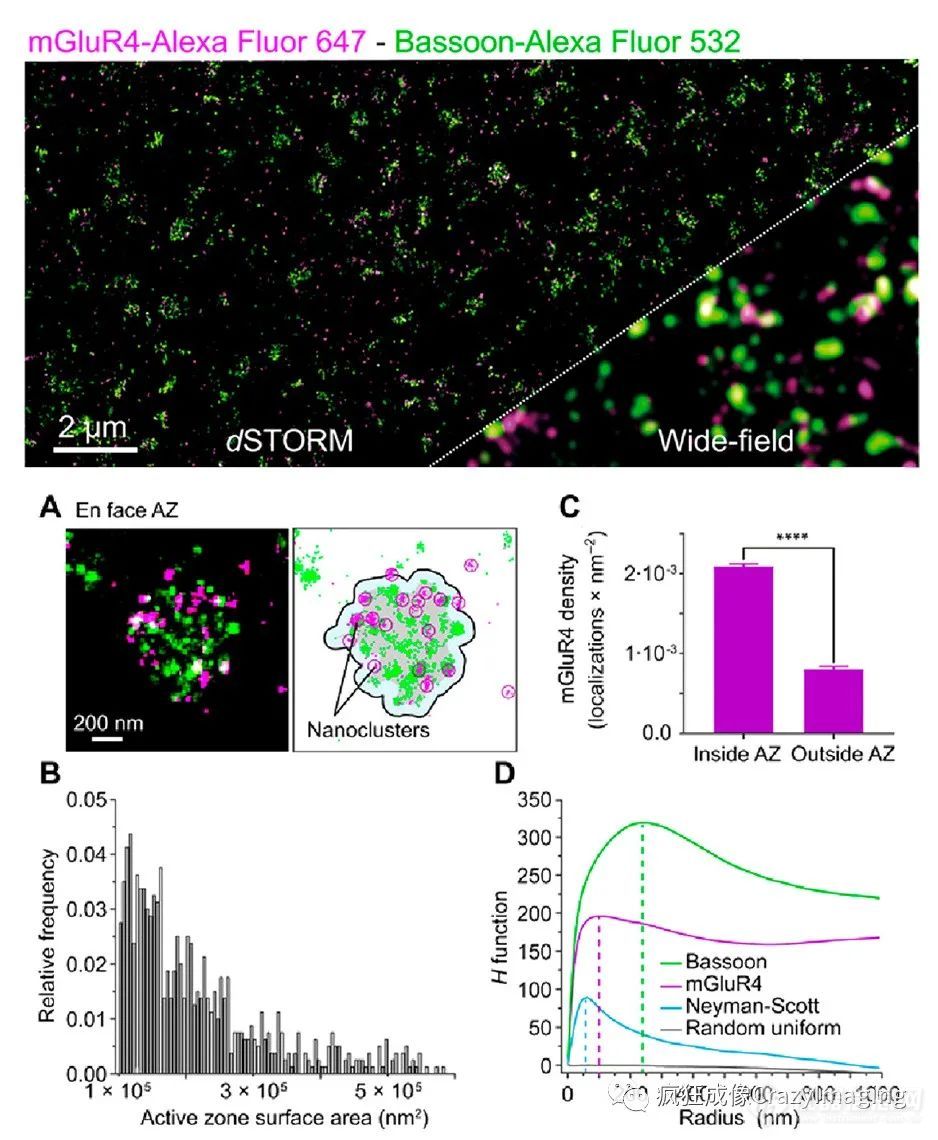
图3小鼠脑片中代谢型mGluR4受体定位定量双色dSTORM。上图:mGluR4和Bassoon免疫染色的小脑冠状切片的dSTORM图像,作为活性区参考。与宽场显微镜结果的比较。(A)DBSCAN聚类算法定义了近距离的En face活性区表面积(灰色)和mGluR4信号(品红)。(B)活性区大小的频率分布直方图(C)mGluR4信号到突触和突触外区域的映射。(D)通过Ripley H函数分析评估Bassoon和mGluR4的聚集分布。与随机分布的分子(蓝色、灰色)进行比较。虚线表示Ripley分析的最大值。
这些研究显示了定量SMLM在神经科学研究中的潜力。可以预见,定量SMLM的进一步发展将为突触前和突触后蛋白质的功能关系,及其组织和结构的研究提供更有价值的信息。
2.2.5. 组织三维(3D)SMLM
虽然SMLM方法实现了仅几纳米的非常高的水平定位精度,但它需要特殊的方法来打破图像平面上方和下方PSF的对称性,来实现高轴向定位精度。实现高轴向定位精度的两种方法是PSF重塑和多焦面检测,通常用于在3D中精确定位荧光团。在SMLM中最常用的方法是通过在成像路径中插入单个柱面透镜从而不对称地扭曲PSF,利用光学像散原理来实现三维定位。基于像散方法的3D dSTORM技术还可以与光谱拆分相结合,对COS-7细胞中的网格蛋白表面小窝成像。像散引起的畸变程度由荧光团的轴向位置决定,因此可用于轴向位置计算。例如,3D散光SMLM已用于确定抑制性突触后密度区gephyrin蛋白和受体复合物的分布和拷贝数,或突触前活动区和突触后密度区各种成分的空间关系。采用双物镜像散成像方案,通过3D SMLM研究组织中肌动蛋白、血影蛋白和其他相关蛋白的结构,发现这些蛋白在轴突中形成190nm的周期性环状结构。替代方法包括使用相位掩模、变形镜实现双螺旋、四足或鞍点PSF重塑,和双焦面成像方法实现更大的轴向范围,并已成功应用于不同的应用中。为了在2D和3D中定位单个荧光发射区,已经开发了不同的算法和软件工具。在最近的一次综述中,列出了不同3D SMLM方法获得的水平和轴向分辨率,以供比较。
然而,到目前为止,大多数SMLM研究都是在培养细胞上进行的。培养细胞具有相对简单的样品制备和成像要求,例如焦平面上下荧光分子的背景信号较低或没有。而脑片更具挑战性,因为它们在焦平面上下表现出高密度的神经元结构和潜在的高荧光背景,由此产生的低信噪比对于成像来说是一个障碍,会直接影响单分子定位的精度和准确性。此外,脑片必须具有足够的渗透性,以实现有效的免疫标记,而不损害组织结构。除了超微结构保存外,对几十微米厚的脑片进行均匀标记也仍然是一个挑战。因此,最初的尝试是将组织切成薄片,以便于标记。然而,将多个切片进行三维重建也仍然具有挑战性。为了保存好抗原表位,在切割成超薄切片进行SMLM成像之前,需要对组织进行解剖、固定以及免疫标记、后固定、脱水并包埋在环氧树脂中。而且,3D SMLM只能对靠近盖玻片的脑片的一个轴向平面进行成像。如果背景信号和散射太强,也可以使用组织透明化方法和基于光片的照明方法进行SR成像。
另一个导致较厚脑片的3D成像不可靠的原因是光学畸变引起的单个荧光团PSF变形和模糊。通常应用自适应光学恢复PSF,使各神经元能够更精确地成像。为了进一步提高基于自适应光学的SR成像的性能,需要将其与自适应PSF重塑相结合。例如,在30μm厚的阿尔茨海默病小鼠模型脑片中,通过同步校正样品引起的畸变并且生成一致的PSF,可以重建纤维淀粉样蛋白斑块的精细细节。荧光团的3D位置也可通过PSF内产生的自干涉精确确定,并已成功用于轴向深达50μm的 SMLM。
3.膨胀样品显微成像技术(EXM)
2015年,Ed Boyden及其同事描述了另一种绕过荧光显微镜分辨率极限的创新策略,称为膨胀样品显微成像技术(ExM)。ExM的原理是将蛋白质连接到带电荷的聚丙烯酰胺凝胶中,然后使用蛋白酶进行轻微的机械破坏,然后让其在水中膨胀,从而实现样品的物理放大。最初的方法是将三功能连接体交联到凝胶基质上,将标签信息转移到凝胶上。
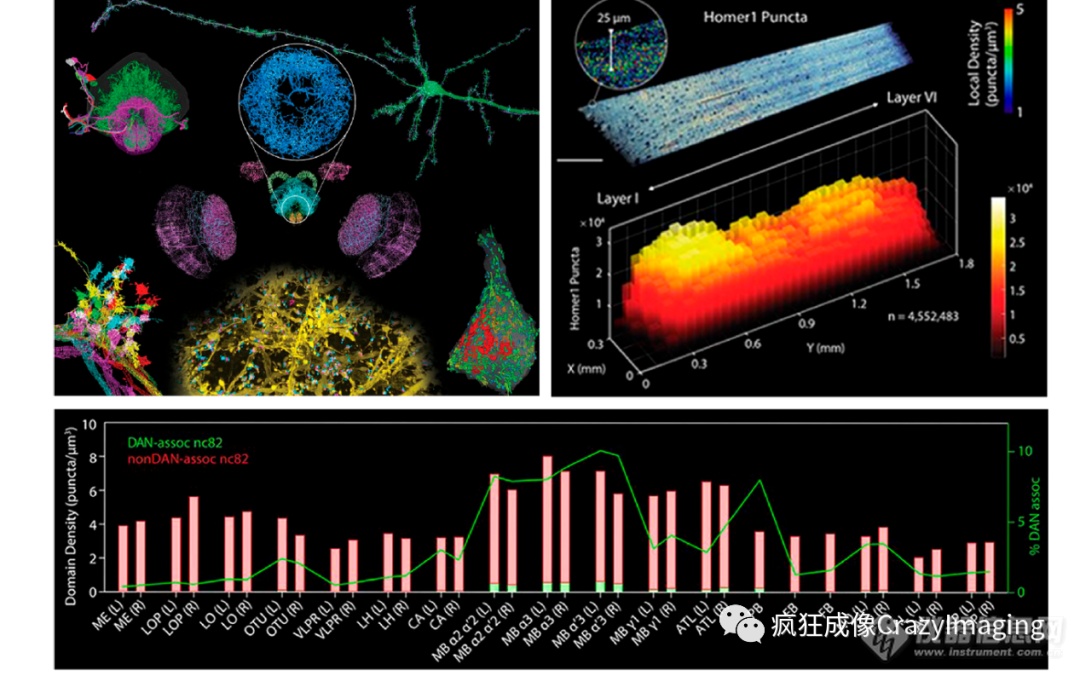
图4 结合ExM和晶格光片显微镜(LLSM)可观察大量神经元分子组装细节。
左上图:概图;右上部位:海马CA1锥体神经元的投射;右下部位:神经元胞体;中下部位:小鼠大脑,通过Thy1 EYFP信号和突触蛋白免疫组织染色进行皮质树突棘成像;左下部位:果蝇投射神经元(PN)束的多样性。左上部位:果蝇中枢复合体PN的神经元追踪。中心部位:果蝇多巴胺能神经元的全脑成像。
下图:成年果蝇大脑所有33个脑区的多巴胺能神经元相关Brp信号(体积密度)定量图(nc82抗体免疫染色,绿色曲线=与多巴胺能神经元相关的nc82斑点百分比)。
右上图:沿着皮质层I至VI(顶部,最小强度投影)的初级躯体感觉皮层中的Homer 1点密度和25μm×50μm×50μm跨皮质的Homer 1点累积数量。请注意,在第二层/第三层和第五层中,Homer 1的密度较高。
三功能连接体含有甲基丙烯酰基团、荧光标记和寡核苷酸,它与连到蛋白质标记的抗体的互补寡核苷酸杂交。将免疫染色的细胞或组织包埋在单体溶液中后,添加含有丙烯酸钠(凝胶高吸水性树脂)的共聚单体丙烯酰胺和交联剂N,N′-亚甲基双(丙烯酰胺)。这些单体组分和位于目标上的甲基丙烯基在高温下聚合,使用四甲基乙二胺(TEMED)作为催化剂,过硫酸铵(APS)作为聚合引发剂。
荧光标签通过锚定在凝胶中的特定位置而在空间中固定,并且其位置可以在蛋白酶的化学预处理和水中透析后延伸构象进行物理膨胀。最初使用ExM产生了一个100倍的体积膨胀和目标分子间的4−5倍距离的线性增加。使用常规共聚焦显微镜对其成像,SRM横向分辨率可达到70 nm,轴向分辨率可达200 nm。在随后的几年中,通过使用交联分子如MA-NHS和acryloyl-X将蛋白质锚定到凝胶基质上,开发了改进proExM方法,使用常规荧光团就可保留蛋白质表位。为了避免自由基起始剂对荧光团的影响,设计了新的方法如蛋白质组放大分析(MAP),与SDS结合热介导变性,实现膨胀凝胶的后标记。
此外,通过引入三功能连接体,它们能够在聚合、消化和变性后存活,并能够将目标分子和官能团直接共价锚定到水凝胶中,以实现膜和细胞骨架的SR成像。有研究显示,一些荧光团与激动剂发生反应。例如,因为菁染料在样品制备过程中几乎消失,因此不适合用于ExM标记,而其他荧光团如Alexa Fluor 488、CF和Atto染料在ExM处理流程后仍然能发射足够的光子。此外,生物素−亲和素信号放大和交换反应信号放大免疫染色技术(Immuno-SABER)可以有效提高信噪比和对大量目标蛋白进行成像。最近的研究表明,可以通过优化Ultra(U-ExM)的流程保留中心粒的超微结构细节。
U-ExM启发于MAP方法,该方法允许膨胀并使用低甲醛和丙烯酰胺浓度交联蛋白质(保留样品的超微结构细节)后进行标记。U-ExM已经成功地用于揭示以前只有电镜才能获得的中心粒的超微结构细节。通过使用不同的单体和引发剂组合,Truckenbrodt及其同事设计了一个对常规培养细胞甚至神经元进行10倍体积膨胀的方案,即通过二次凝胶进行迭代ExM制样,多次嵌入不同的单体(而这些单体又可以通过使用高摩尔氢氧化钠去除)从而达到高达53倍的膨胀系数。应用此方法,可以从突触后支架蛋白中清晰看到位于突触间隙内的膨胀13倍的蛋白,例如神经递质受体GluR1和GABAARα1/α2。
如今,ExM已成功用于培养细胞、原代神经元和组织中的蛋白质、RNA、真菌、病理标本和细菌的超分辨率成像。4倍膨胀的ExM已经与晶格光片显微镜相结合,以60×60×90 nm的分辨率对整个果蝇大脑中多种蛋白质之间的纳米级结构进行成像(图4)。
为了进一步提高分辨率,ExM已成功地与SRM方法相结合。例如,ExM与SIM相结合到达20nm的空间分辨率,观察果蝇中的联会复合体和小鼠精母细胞。在细胞培养样品中进行多表位标记后,与传统荧光显微镜相比,使用STED结合ExM可使分辨率提高30倍。
此外,使用NHS染料对所有蛋白进行标记,然后进行迭代ExM,可以对高蛋白密度的结构或细胞器(如线粒体),实现与EM相比具有更高对比度的超微结构细节。
为了在分子尺度上进行成像,ExM与SMLM方法(如dSTORM)相结合是一个理想的选择。然而在含有硫醇和盐的传统光转换缓冲液中,会发生荷电氢凝胶收缩。可通过使用低离子强度缓冲液或加入中性溶液使凝胶稳定以避免收缩。
另一种策略是使用自发闪烁的荧光团(如HMSiR)在水中进行SMLM。通过Ex-dSTORM实现分子分辨率的关键是膨胀后标记,这增加了表位可及性,从而提高了标记效率并减少了标记错误。Ex-dSTORM超分辨成像已成功应用于原代细胞和神经元中微管和中心粒结构的解析。
来源于:疯狂成像Crazymaging












热门评论
最新资讯
新闻专题
更多推荐
