
基于离子淌度质谱对完整蛋白质形态进行非标记定量


导读:随着质谱技术的发展,自上而下蛋白质组学质谱研究中蛋白质的鉴定数量大大提升,生成了包含数万种proteoforms的数据集,但在proteoforms的量化能力没有得到相应的性能提升。
大家好,本周为大家分享一篇发表在Analytical Chemistry上的文章,Improved Label-Free Quantification of Intact Proteoforms Using Field Asymmetric Ion Mobility Spectrometry [1],文章的通讯作者是美国俄克拉荷马大学的Luca Fornelli教授。完整proteoforms的非标记高通量定量方法的应用对象通常为从整个细胞或组织裂解物中提取的0 - 30 kDa质量范围内蛋白质。
然而当前,即使通过高效液相色谱或毛细管电泳实现了proteoforms的高分辨率分离,可鉴定和定量的proteoforms的数量也不可避免地受到固有的样品复杂性的限制。近年来,随着质谱技术的发展,自上而下蛋白质组学质谱(top-down proteomics)研究中蛋白质的鉴定数量大大提升,生成了包含数万种proteoforms的数据集,但在proteoforms的量化能力方面并没有得到相应的性能提升。
为克服这一问题,本文中作者通过应用场不对称离子迁移谱法(Field asymmetric ion mobility spectrometry, FAIMS)对大肠杆菌中的proteoforms进行了非标记定量。由此产生的改进允许在单次LC-MS实验中采用多个FAIMS补偿电压(Compensation voltages, C.V.),而不会增加整个数据采集周期。与传统的非标记定量实验相比,FAIMS的应用在不影响定量准确性的情况下,大大增加了鉴定和定量的proteoforms数量。首先,作者优化了质谱stepped-C.V.数据采集方法对Orbitrap Eclipse性能的影响,并从中筛选出了最优条件(−40、−20、0 V组合)。
所有最新的基于Orbitrap的质谱仪(包括Exploris platform和Orbitrap Ascend)都可以采用single time-domain transients(即单次微扫描)在top down FTMS实验中生成高质量的质谱图。作者认为这对于在单次LC - MS2运行期间应用多个C.V.值的采集策略特别有益。接下来,作者应用该方法对大肠杆菌中的蛋白质进行了检测,并与传统的LC - MS2 DDA采集方法进行了比较(图1)。如图所示,每个C.V.值下的总离子流图都不同,且这一额外的分离导致在LB(Luria broth)和M9(醋酸钠处理)样品中鉴定到的proteoforms的数量显著提升。
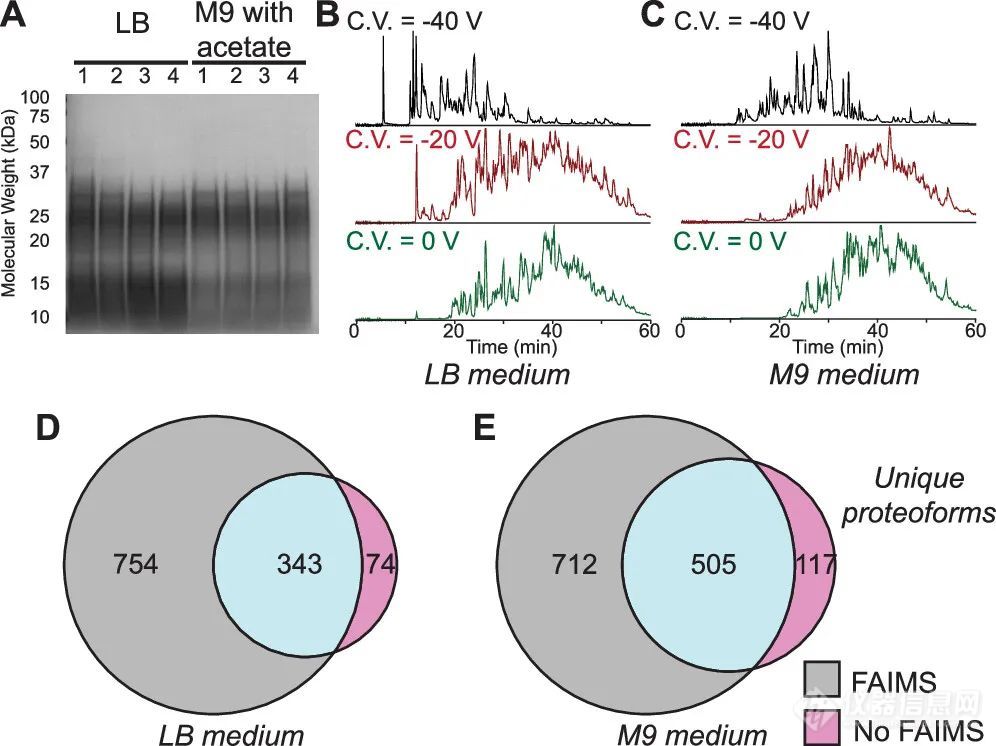
图1. 样本制备方法和proteoforms鉴定结果总结虽然在LC-FAIMS和LC-only数据集中,大多数鉴定到的proteoforms质量都小于15 kDa,但其中约20%的质量大于18 kDa甚至高达33.3 kDa(图2)。对已鉴定的proteoforms列表的深入分析表明,达到鉴定低丰度proteoforms的关键参数之一是在串联质谱(MS2)中有足够的时间注入离子。
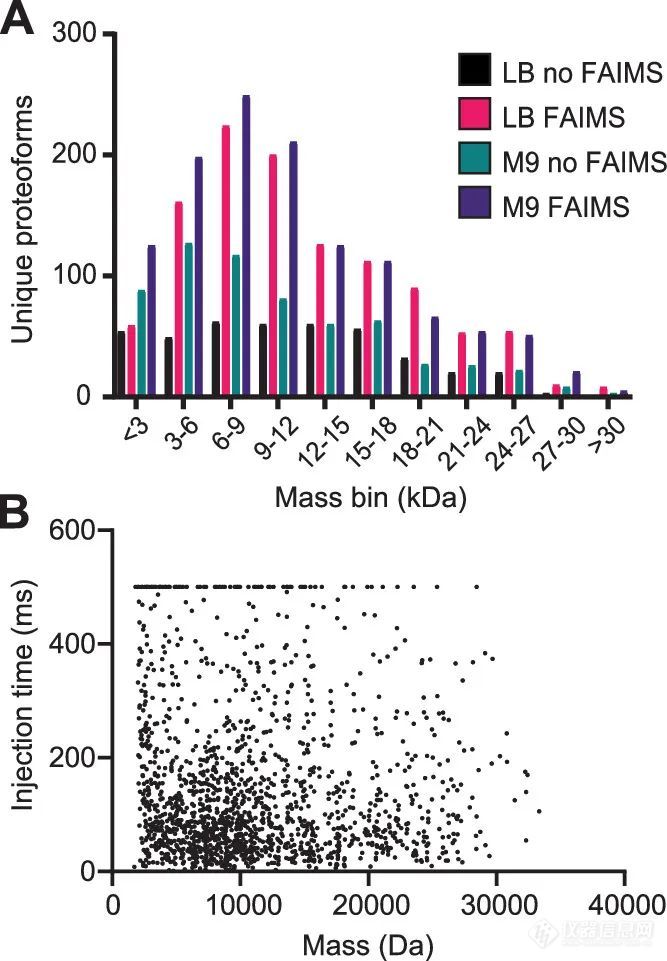
图2. A. FAIMS和非FAIMS鉴定到的proteoforms的质量分布。B. 鉴定到的proteoforms与注射时间之间的关系。最后,作者采用ProSight PD v 4.2 (Proteineous, Inc)进行了基于MS1的非标定量,结果显示基于FAIMS的数据集在LB样品(蓝色)和M9样品中检测到的差异表达的proteoforms均有所增加(图3)。作者评估了两个数据集之间的差异(使用和不使用FAIMS采集数据),以验证FAIMS的应用是否会对量化准确性产生不利影响,结果只有1个proteoform显示相互矛盾的丰度趋势。这种差异是由于该蛋白和一个共流出蛋白之间质谱峰几乎完全重叠造成的。它们具有非常接近的单同位素质量,这样高水平的信号干扰可以很容易地干扰基于MS1的量化。启用FAIMS可以使MS1谱图简化,因为两种proteoforms可以富集在两种不同的C.V. 值下。
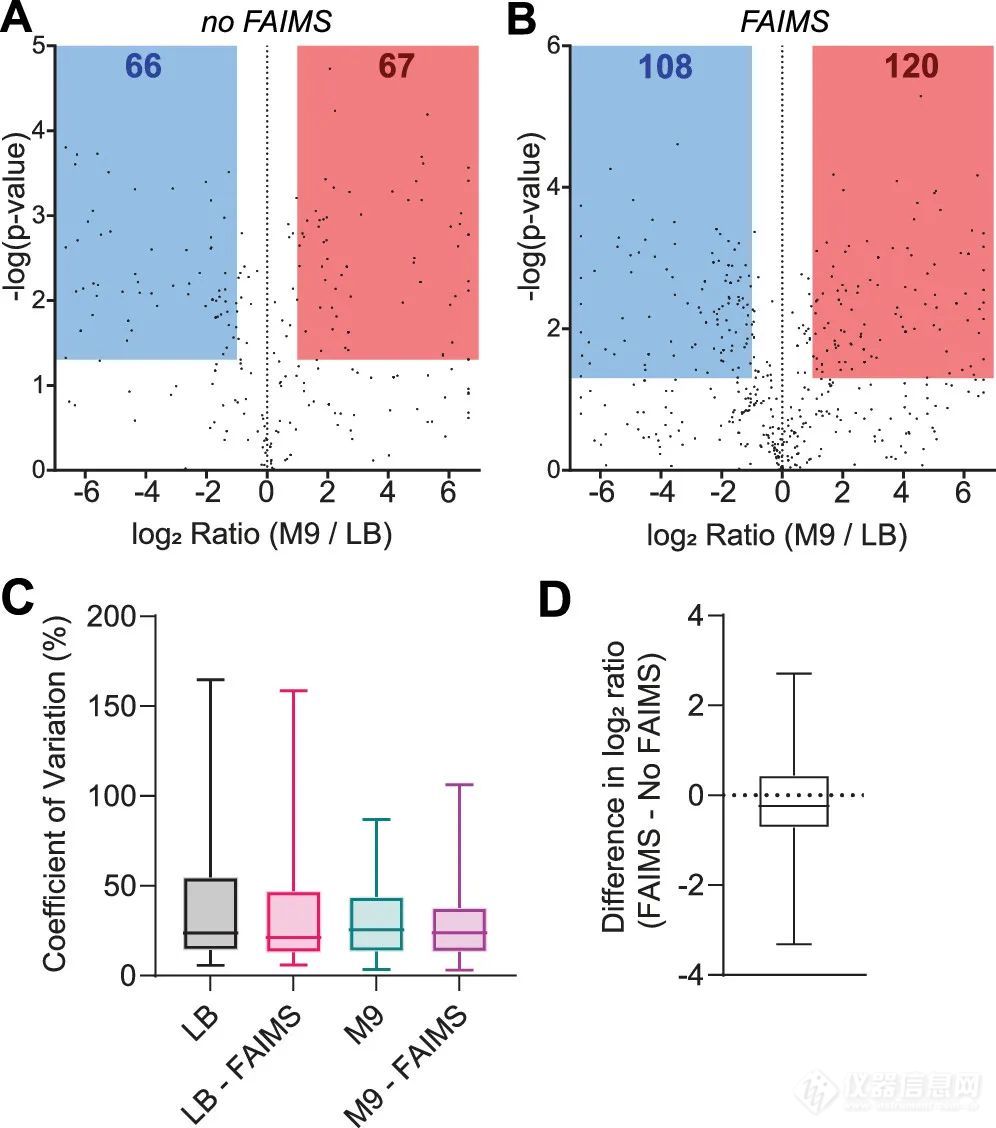
图3. 大肠杆菌proteoforms无标记定量结果分析。作者将LC - FAIMS - MS2数据集与通过BUP在类似样品上获得的非标定量结果进行比较,得出两个主要的结论:1. BUP仍然对蛋白质组提供了更深层次的定量表征; 2. BUP提供了与单个基因相关的所有产物的整体丰度水平信息;而TDP方法表明,给定的UniProt accession可以由多个差异表达的proteoforms组成,可能具有不同的行为(即在给定条件下,一些被上调,另一些被下调)。这一额外的信息可能具有潜在的生物学意义,但在基于BUP的定量分析中可能会被遗漏。本文描述的基于FAIMS的定量数据采集方法与PEPPI(Passively eluting proteins from polyacrylamide gels as intact species)蛋白分离技术完全兼容,产生0 - 30 kDa的组分,并且可以方便地根据待分析蛋白的平均质量调整质谱参数(C.V.值),未来在更大的蛋白质定量方面具有广阔的应用前景。
撰稿:张颖
编辑:李惠琳
原文:Kline JT, Belford MW, Huang J, Greer JB, Bergen D, Fellers RT, Greer SM, Horn DM, Zabrouskov V, Huguet R, Boeser CL, Durbin KR, Fornelli L. Improved Label-Free Quantification of Intact Proteoforms Using Field Asymmetric Ion Mobility Spectrometry. Anal Chem. 2023 Jun 13;95(23):9090-9096.
李惠琳课题组网址www.x-mol.com/groups/li_huilin
参考文献
1.Kline JT, Belford MW, Huang J, Greer JB, Bergen D, Fellers RT, Greer SM, Horn DM, Zabrouskov V, Huguet R, Boeser CL, Durbin KR, Fornelli L. Improved Label-Free Quantification of Intact Proteoforms Using Field Asymmetric Ion Mobility Spectrometry. Anal Chem. 2023 Jun 13;95(23):9090-9096.
来源于:仪器信息网














热门评论
最新资讯
新闻专题
更多推荐
