
使用非数据依赖采集法实现氢/氘交换质谱数据自动化分析
导读:本文使用一种自动化HDX数据分析的方法,利用DIA采集方法同时从MS1和MS2领域获取氘代数据,并开发了AutoHX软件来挖掘和分析HDX数据。
HDX-MS是一种基于蛋白质主链酰胺氢原子与氘水中氘原子交换而获取有关蛋白质高阶结构和动态信息的方法。该技术可以帮助研究蛋白质折叠机制、发现配体结合位点、突出变构效应,在生物医药行业中发挥重要作用。尽管HDX-MS在蛋白质分析中频繁使用,但它通常无法进行高通量分析,且受限于大于150 kDa蛋白的分析。此外,HDX-MS生成复杂的同位素峰型常伴有谱图重叠现象,导致氘代值被错误计算。随着样品复杂性的增加,这一问题会更加加剧。目前,数据处理的方法涉及到手动检查原始数据以筛选谱图,并丢弃有任何信号问题的肽段图谱。然而这种方法随着样品分子量和复杂程度的增加变得难以执行,且容易受到人为错误的干扰(图1)。因此迫切需要一种可以消除手动筛选数据的负担,同时能够兼容更复杂的谱图(来自复杂混合物或整个细胞裂解液样品的谱图)。本文作者使用了一种自动化HDX数据分析的方法,利用data independent acquisition(DIA)采集方法同时从MS1和MS2领域获取氘代数据,并开发了AutoHX软件来挖掘和分析HDX数据。
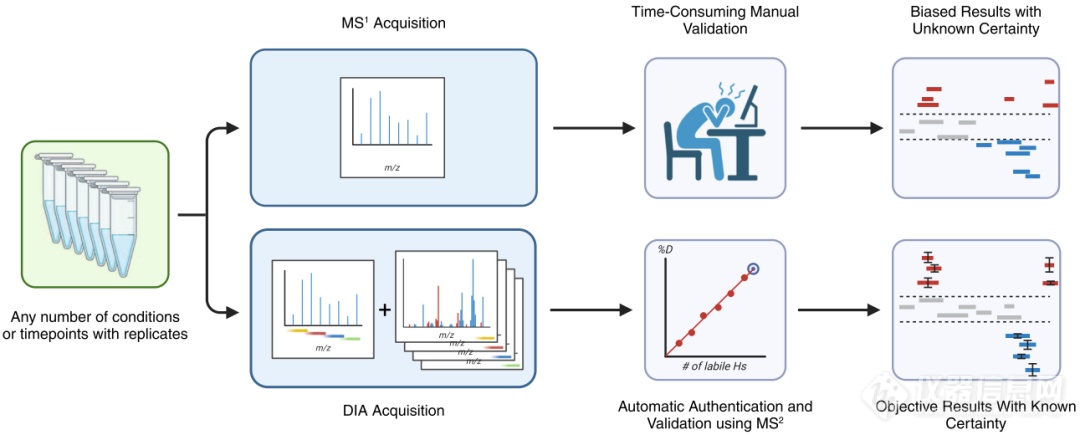
图1.传统HDX-MS数据采集与分析流程和本文使用的数据采集和分析流程比较。
针对使用HDX-MS时,碰撞诱导解离(CID)碎裂模式产生的肽段碎片会伴随着气相中的氘重组现象(即scrambling现象),会影响残基水平氘代值的准确测量这一问题,作者定量研究了HDX-MS2数据的特性。作者发现,scrambling与离子传输和碎裂能量有关,且在高传输效率的条件下scrambling较严重,因此首先使用较为温和的离子传输参数和碎裂能量能够降低scrambling程度。随后作者建立了可描述碎片氘代值与该肽段可碎裂位点数量之间的线性关系(图2)。随着碎片离子长度的增加,相应的碎片离子氘代值会线性增加,因此通过回归计算可以计算出整个肽段的氘代率。这种方法不仅利用了CID产生的碎片信息,同时更为准确的计算出肽段的氘代值,排除了肽段谱图重叠对计算氘代值的干扰。
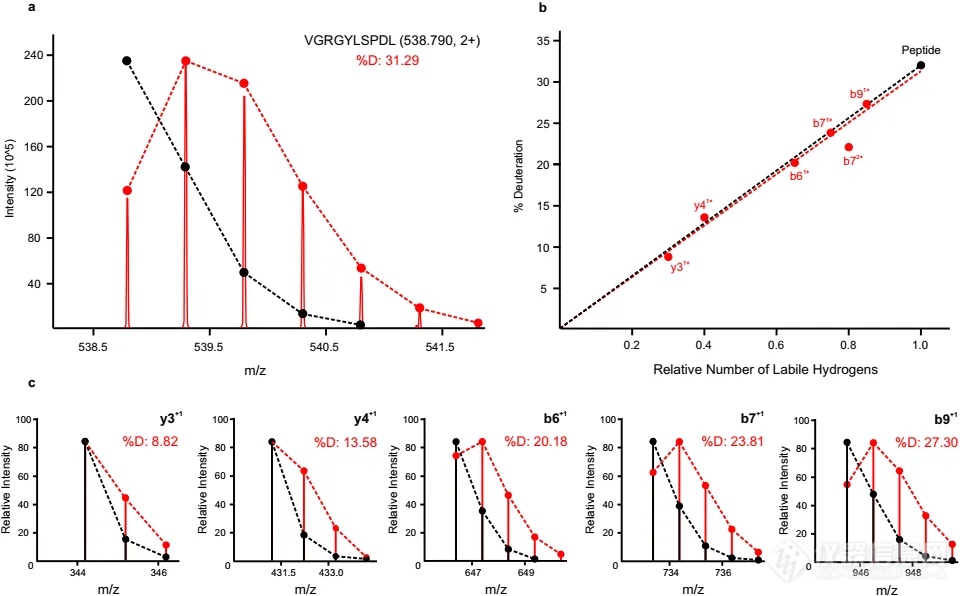
图2.在一条给定肽段中,HD scrambling中,氘代值与碎片长度的关系。
接着作者提出使用DIA方法来获取HX-MS2实验中MS1和MS2域的氘化数据,以实现在不同质谱平台采集数据、采集复杂样品的信息、分析自动化数据,且使得通过CID产生的MS2中提取肽段氘代值成为可能。首先作者设置了尽可能小的DIA窗口,并使用了较大的窗口重叠区域,以最小化MS2谱图的复杂性并确保每条氘代肽段至少有一个窗口(图3)。同时,作者开发了一个名为AutoHX的软件(作为Mass Spec Studio中的插件),该软件自动选择理想的DIA窗口,并从MS1数据计算前体肽段的氘代值,以及从MS2数据计算所有碎片的氘代值。同时改进了HX-PIPE(为HDX-MS量身定制的搜索引擎),使其搜库结果直接应用于AutoHX的分析。随后AutoHX使用了一系列过滤器来从数据集中解析低质量信号,然后使用基于RANSAC的谱图分析器,为所有肽段及其碎片匹配最佳同位素集合,并绘制动力学曲线图。该方法显著提高了肽段序列覆盖的冗余度(图4),从而提高了测量质量。
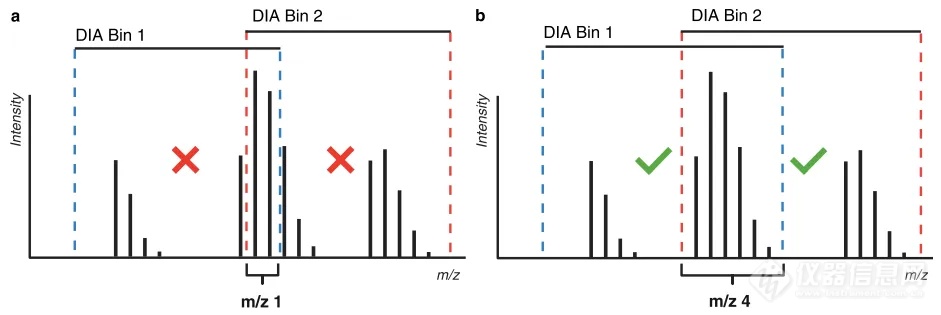
图3. DIA窗口设计示意。
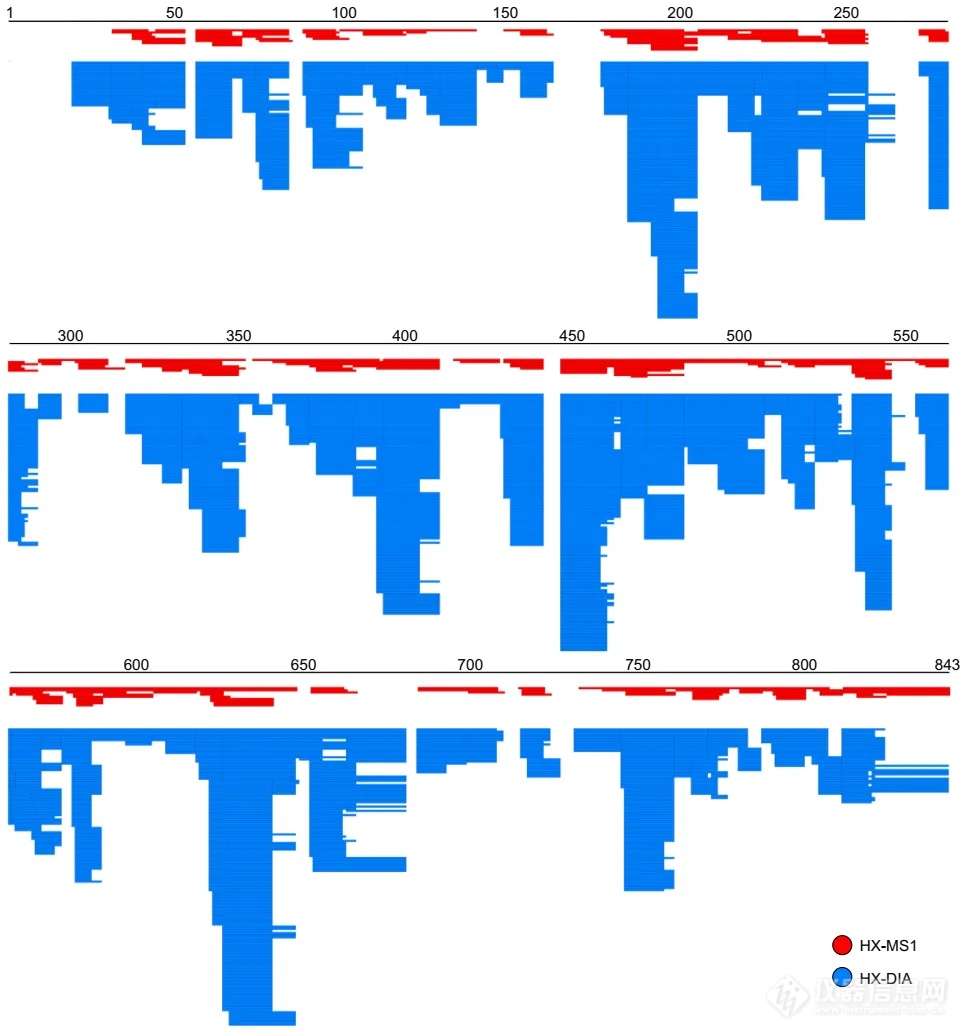
图4. 基于DIA采集模式得到的序列覆盖(糖原磷酸化酶B,phosphorylase B)与基于传统HDX-MS中MS1采集模式的结果比对。
接着,软件会通过MS1和MS2数据收集到的肽段前体离子和肽段碎片离子的信息,计算出相应的氘代值,同时将所有重复组计算出的氘化值集合成一个分布(通常为正态分布),并从该正态分布中,选择最接近平均值的组合,即为精确的氘代值,利用每个时间点的氘代值生成HDX动力学曲线(图5)。作者将手动筛选检查的数据与自动分析法获得的氘代数据进行了比对,结果具有一致性,验证了自动化方法的准确性和可靠性(图6)。同时在做同一样本不同状态HDX比较实验时,AutoHX可以生成氘代差异的显著性差异分析图(Woods plot)(图7),用于比较不同状态下的蛋白结构和构象差异。
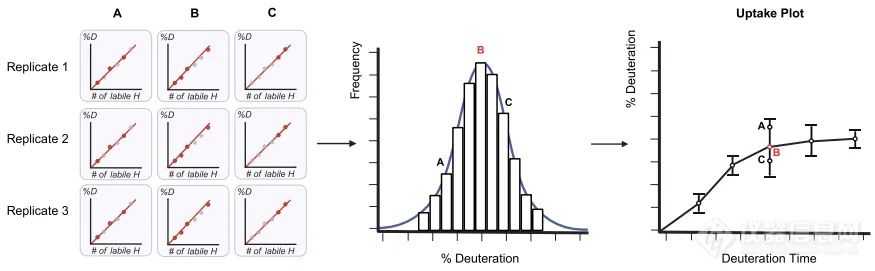
图5. 氘代曲线的组合方式。
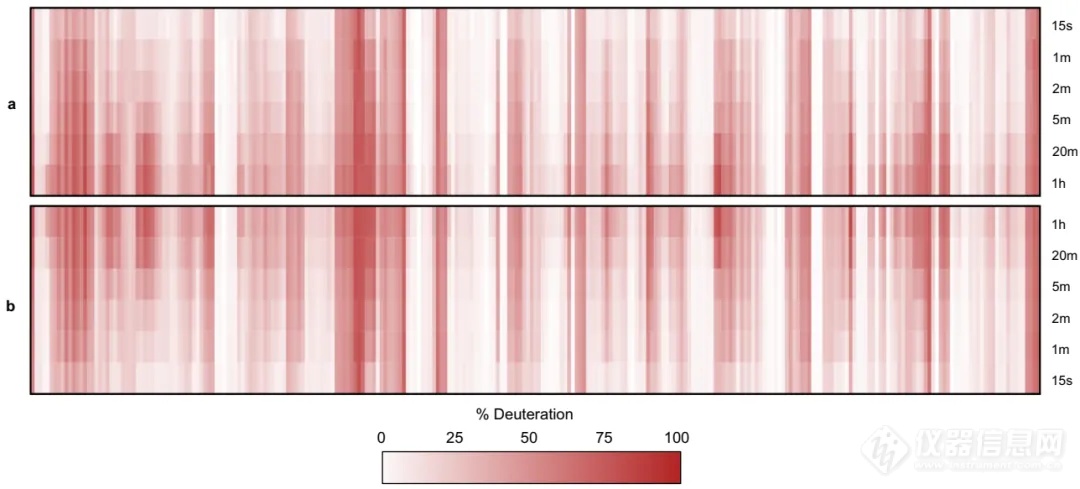
图6.手动MS1数据分析和AutoHX自动计算的氘代率对比。
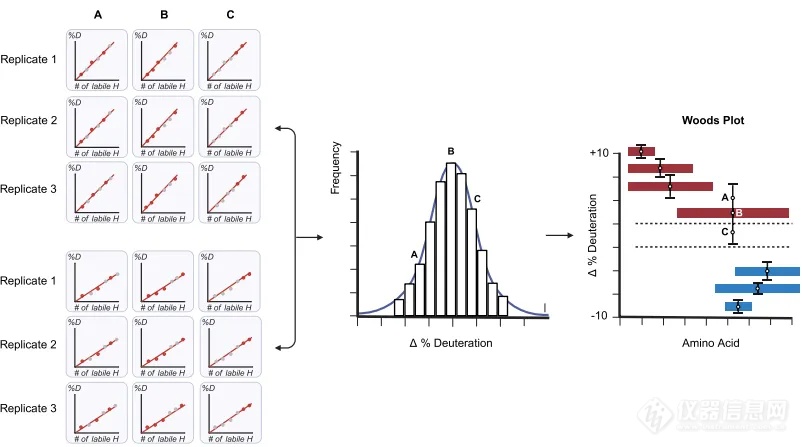
图7.氘代差异分析流程示意图。
最后作者用两个蛋白体系验证了该方法的实用性和可靠性。第一个体系为DNA聚合酶ϴ(Pol ϴ)与其抗生素药物novobiocin结合的结构变化。通过比较手动处理与自动化处理的数据,作者发现生成的氘代差异图结果相似,提示该方法具有较好的准确性,并能够定位结合带来的氘代上升和下降区域(图8)。第二个体系是DNA依赖性蛋白激酶(DNA-PKcs)与选择性抑制剂AZD7648的结合。使用AutoHX软件处理了六个HDX-MS实验的数据,快速生成了Woods图,发现大部分可检测到的稳定性增加集中在FAT和激酶结构域(图9b),还包括药物结合位点的铰链环区域(图9c),揭示了药物结合位点及其引起的动态性变化。这部分研究结果展示了自动化数据分析在药物结合研究中的有效性,特别是在分析大型蛋白质复合物和难以纯化的蛋白质时,为药物开发和疾病治疗提供了有价值的信息。
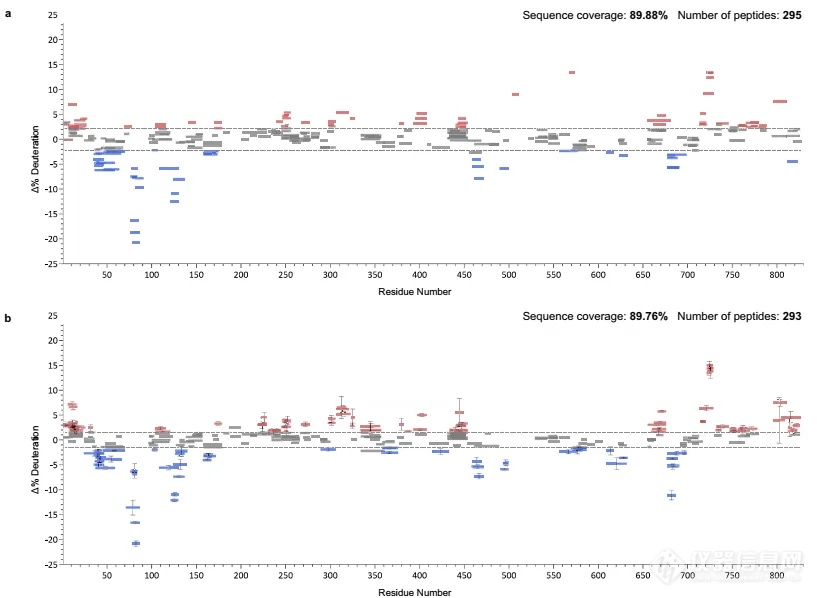
图8.手动处理与自动处理的Pol ϴ与novobiocin-bound Pol ϴ的HDX数据作差对比。
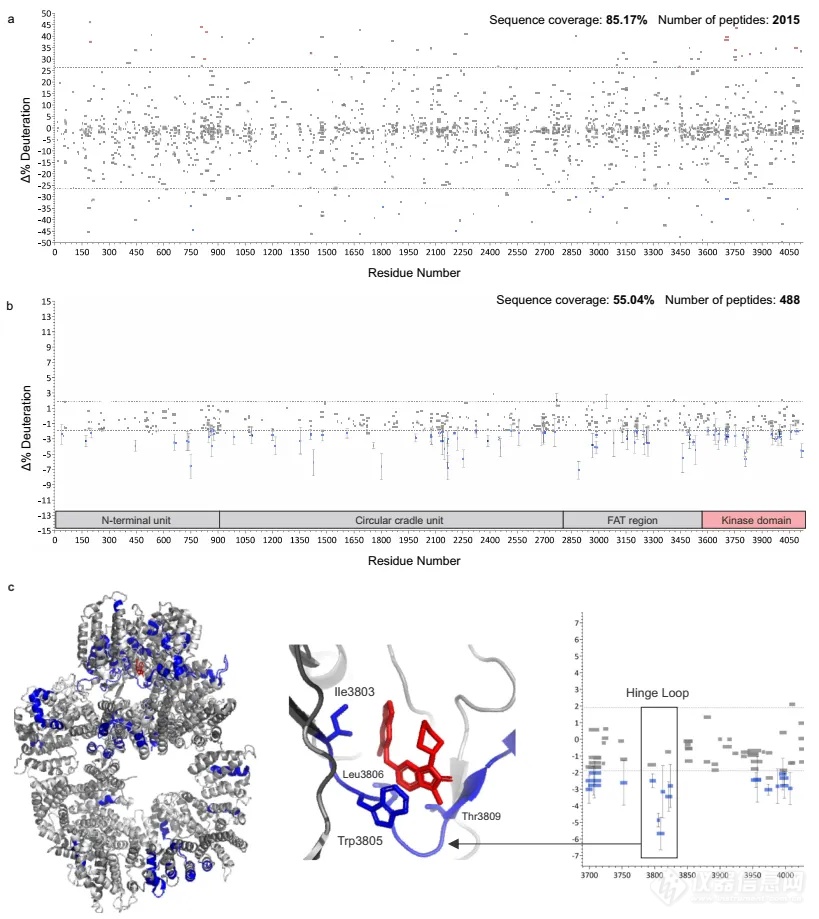
图9. DNA-PKcs+AZD7648的自动化HDX分析流程结果。
总的来说,该研究开发了AutoHX软件,通过自动化数据分析和基于DIA的HX-MS2工作流程,显著提高了氢/氘交换质谱技术在蛋白质结构和药物结合分析中的效率与应用范围,使得这一领域技术更加易于使用并可供更广泛的科研社区应用。该工作的亮点,从实验设计上:考虑到了目前HDX-MS流程——数据采集、数据分析——中存在的瓶颈与局限。从方法学考察层面:方法验证科学严谨、周到。从技术上:大大降低了人工处理HDX-MS数据的成本,提高了检测能力,有提高检测通量的潜力。从科学思维上:利用了scrambling的规律,将普遍的问题转化成了机遇。HX-DIA提供了一个概念上的转变,降低了该技术的使用门槛,使该技术“平民化”。
本文发表在Nat. Commun.上,题目为“Automating data analysis for hydrogen/deuterium exchange mass spectrometry using data-independent acquisition methodology”,作者是加拿大卡尔加里大学的David C. Schriemer。
来源于:仪器信息网


李惠琳课题组
总阅读量 0







近期会议
更多




热门评论
最新资讯
新闻专题
