杨幸怡
南方医科大学
研究背景:苯丙胺类药物(Amphetamine, AMPH)是一类成瘾性药物,使用这类药物可以让人产生欣感、厌食以及各种幻想。AMPH类药物的核心结构都是p苯乙胺,这类药物都有容易通过血脑屏障,抵御生物转化以及刺激神经末梢释放单胺类递质的特点。MPH类药物还可以通过抑制单胺氧化酶(monoamine oxidase,MAO)对胺类物质的氧化。甲基苯丙胺(Methamphetamine, METH),俗称“毒,是新型合成类毒,属于苯丙胺类神经兴奋剂(Amphetamine-Typed Stimulant, ATS),具有见效快、兴奋作用维持时间长,价格低廉,化学合成技术简单,多途径摄取等特点,成为全球第二个使用最广泛的非法药物类别。吸服毒后容易造成一系列不良症状,例如心血管疾病、精神病、神经精神症状、脱水、横纹肌溶解以及肝肾衰竭,这些不但给吸毒个人带来痛苦的体验,更给家庭以及社会带来沉重的负担。METH最大的特点是一旦使用便会上瘾。它的另一特点,是在成瘾的同时会对人体重要器官造成器质性损害,特别是对中枢神经细胞的毒性损伤作用与其他毒相比,异常突出。METH可导致人大脑黑质、纹状体、海马、皮质等多部位损伤,星状细胞病变、脑肿胀和变性、苍白球的退行性变化、脊髓灰质的坏死等。研究报道,METH滥用者有脑结构异常,表现为纹状体体积增大,海马体积减少,白质体积增大,大脑皮质体积减小,选择性的内侧颞叶和扣带-边缘区域损害等。这些部位与大脑代谢失调、记忆障碍等相关、认知障碍、警觉功能障碍有关。临床上METH滥用者出现记忆力下降、幻听、被害妄想、易激惹、性暴力、自杀、他杀等精神和暴力行为,与上述改变密切相关。已证实,METH滥用所引起的刑事犯罪更为突出,不同的是海洛因成瘾者的犯罪多为筹措毒资,而METH滥用的刑事犯罪主要是在毒性作用下的直接暴力行为所致。这提示METH滥用所致的毒性损伤作用强,对社会治安的危害性更大。关于METH是如何导致上述损伤,其机制仍不明确,现阶段的研究结果显示多种机制参与了METH的神经毒性,主要包括氧化应激、神经元凋亡、兴奋性毒性、线粒体功能障碍等,其中氧化应激是METH所致神经毒性损伤的重要机制作用之一。其中这些机制并不是独立存在,既有相互交叉,又分别发挥着不同的作用。我们对METH导致的神经损伤大体分为两类,一类是导致氧自由基、氮自由基等活性物质的清除能力减弱和导致的神经损伤,属于防护能力不足。另一类是损伤相关通路的层层激活以及细胞器的受损又推动神经损伤的进一步加剧,包括一些凋亡通路的正性激活。过去我们的研究发现METH使用使得多个脑区激活nNOS/NO,促进生成大量的硝基化蛋白。谷胱甘肽S-转移酶1(Glutathione S-transferase Pi, GSTP1)在毒理学上有一定的重要性,它可以催化亲核性的谷胱甘肽与各种亲电子外源化学物的结合反应。许多外源化学物在生物转化第一相反应中极易形成某些生物活性中间产物,它们可与细胞生物大分子重要成分发生共价结合,对机体造成损害。谷胱甘肽与其结合后,可防止发生此种共价结合,起到解毒作用。而GSTP1的硝基化可能会导致到对催化GSH结合亲电子外源化学物的能力减弱从而导致GSH抵抗氧化应激的能力减弱。近年来发现GSTP1是调节CDK5的新基因,GSTP1可通过直接解除P25/P35的结合去抑制CDK5的活性,还可以间接消除氧化应激带来的损伤,减少CDK5激活所带来的神经退行性变以及神经细胞的死亡[13]。METH的神经毒性还表现在对细胞骨架的损伤,这成为了成瘾以及运动功能异常的结构基础。我们前期体外细胞实验中发现Rho相关激酶2(Rho-associated coiled coil forming protein kinase Ⅱ, ROCK2)在METH处理后表达增加活性增强。过去的研究中认为ROCK2参与了生长锥的坍塌以及神经元的的回缩。ROCK的抑制可以保护缺血导致的脑部损伤,防止神经元的坏死与凋亡。于是本课题第二部分实验提出了抑制ROCK2可能对METH损伤的神经系统有保护作用是第二章要解决的内容。抑制ROCK2对细胞骨架以及神经元的损伤是否有保护作用;ROCK2在METH导致的神经毒性中扮演的角色以及相关机制是本论文重点讨论的问题。上述两部分的研究目的在于探寻METH导致神经毒性的机理以及寻找合理的干预手段。方法:第一章第一节建立7-NI抑制nNOS/NO的METH中毒体内模型,通过western blot以及免疫组化观察GSTP1在METH以及抑制1iNOS/NO抑制后的表达情况以及3-NT/GSTP1的比例。第一章第二节构建GSTP1过表达慢病毒,通过免疫荧光以及MTT选择合适的病毒感染滴度,通过Western blot验证LV-3flag-eGFP-GSTP1在PC12以及原代黑质神经元中的转染效率;通过Annexin V-PE/PI双染以及Hoechst染色检测过表达GSTP1的PC12细胞以及原代黑质神经元与野生型PC12细胞以及原代黑质神经元在METH处理后凋亡率的改变。Western blot验证过表达GSTP1对CDK5在METH处理后表达量的影响。通过化学检测法检测GSTP1过表达对METH导致ROS产生量的影响。第一章第三节通过在双侧SN区立体定位注射GSTP1重组慢病毒,通过荧光显微镜以及Western blot观察其在SN区以及各投射的脑区的表达情况。第二章第一节通过QPCR以及Western观察ROCK2在PC12细胞中的增高是否具有METH浓度依赖性。构建ROCK2特异性小RNA干扰siROCK2,通过western blot、QPCR以及流式细胞技术检测siROCK2在PC12细胞中的转染率以及干扰率。通过流式细胞检测凋亡技术Annexin V-FITC/PI双染以及TUNEL染色检测干扰ROCK2对METH导致PC12细胞的凋亡作用。通过化学检测检法检测ROCK2抑制对METH导致ROS产生量的影响。通过QPCR检测RhoA在METH处理PC12中转录水平的改变,以及干扰ROCK2是否对RhoA的转录水平产生影响。通过Western blot检测干扰ROCK2对METH相关的Akt磷酸化、caspase-3活化以及PARP分裂的作用,探寻抑制ROCK2逆转METH导致神经细胞凋亡的相关机制。通过免疫荧光染色观察ROCK2干扰对METH导致原代黑质神经元细胞骨架、树突棘密度以及树突长度以及树突分叉的改变。通过western blot检测干扰ROCK2是否对METH处理原代黑质区神经元的MLC的磷酸起作用,探寻抑制ROCK2正性调节METH导致细胞骨架异常的机制。第二章第二节通过western blot以及免疫荧光观察METH处理后PFc、Str、Hip以及SN中ROCK2表达的改变。使用ROCK2药物阻断剂Fasudil建立ROCK2干扰的METH体内亚急性动物模型,通过Open field观察Fasudil对METH毒性损伤相关自主运动功能是否有逆转功能。通过western blot检测Fasudil对METH处理后大鼠脑组织中ROCK2降低的量。通过TUNEL以及尼氏染色观察METH亚急性中毒模型中神经元的损伤,TH染色观察METH处理后在Str以及SN两个脑区中TH-ir神经末梢以及胞体的改变,判断Fasudil对METH损伤的神经细胞有保护作用.通过免疫组化集合比较Fasudil是否对METH损伤相关内质网应激标记Grp78、线粒体损伤标记Bax、凋亡下游caspase-3激活态、DAT、以及促进氧化应激因子CDK5在SN区的表达有影响,探寻抑制ROCK2对METH神经性逆转的可能机制。结果:第一章第一节图1-1-1显示nNOD/NO抑制的METH亚急性模型构建成功。图1-1-2A免疫组化显示METH组与空白处理组相比GSTP1在Str以及SN区的表达降低。图1-1-2B Western blot显示在Str区中,7-NI+METH组中3-NT/GSTP1比值比METH组降低58.24%(*p<0.01,n=3);7-NI+METH组GSTP1蛋白含量比METH组增加158.0%(#p<0.001,n=3),图1-1-2C Western blot显示在SN区中,7-NI+METH组中3-NT/GSTP1比值比METH组降低44.3%(*p<0.05,n=3);7-NI+METH组GSTP1蛋白含量比METH组增加了161.0%(#p<0.001,n=3)。第一章第二节图1-2-1至图1-2-6显示成功构建GSTP1重组慢病毒。图1-2-7原代黑质神经元的鉴定结果为TH-ir神经元占该原代培养神经元的31±3%。图1-2-8显示LV-3flag-eGFP-GSTP1转染原代黑质神经元的最佳滴度为5×106TU/ml,图1-2-9显示LV-3flag-eGFP-GSTP1转染PC12中最佳转染滴度为1×107TU/ml。图1-2-10流式检测PC12细胞凋亡显示在最佳LV-3flag-eGFP-GSTP1转染PC12细胞的滴度下,METH处理的情况下,ExpGSTP1组比saline组凋亡率降低66.5%,*p<0.001,n=3。图1-2-11Hoechst标记凋亡显示,在METH处理下,ExpGSTP1比METH组原代黑质神经元Hoechst阳性细胞数降低50.03%(p<0.001,n=3),认为GSTP1的过表达可部分降低METH导致原代黑质神经元的hoechst阳性率。图1-2-12A PCR结果显示2.0mM METH处理PC12细胞24h后,METH组中CDK5mRNA水平与Con组相比升高了157.0%,*p<0.001,n=3,结果说明METH处理增加CDK5的mRNA水平。图1-2-12B Western blot结果显示GSTP1过表达可部分降低METH导致增高的CDK5。图1-2-13显示GSTP1过表达可部分逆转METH导致增多的ROS。在PC12细胞中METH+ExpGSTP1组与单加METH组相比可使ROS减少5.73%,F=40.83,*p<0.05。在原代黑质神经元中METH+ExpGSTP1组与单加METH组相比可使ROS减少6.9%,F=97.88,**p<0.01。第一章第三节图1-3-1结果显示,5uL LV-3flag-eGFP-GSTP1进行SN区立体定位注射8周后,发现SN区以及SN大部分的投射区都带有绿色荧光蛋白,图1-3-2Western blot结果显示PFc、Str、Hip以及SN区出现GSTP1的过表达。第二章第一节图2-1-1A显示METH处理可导致ROCK2的mRNA水平升高,在METH为2.0mM和2.5mM的浓度时,ROCK2mRNA的表达分别为对照组的4.78和4.99倍,具有显著性统计学差异(F=5.05,*P<0.05,n=3)。图2-1-1B、C显示METH处理可导致ROCK2蛋白水平的增加。2.0mM METH处理PC1224h后荧光显微镜镜下观察,METH处理的PC12细胞的突起变短,绿色荧光(ROCK2标记)的强度明显增强。Western blot显示随METH浓度增加160kD位置上的密度增加。图2-1-2A、B、B1显示Lipo作为载体对siROCK2转染PC12细胞的影响,Lipo/siROCK2组的转染效率为69.86%,而siROCK2组的效率和对照组的效率分别为0.66%和0.59%。图2-1-2C显示siROCK2最佳转染时间为48h,图2-1-2D显示siROCK2最佳转染浓度为100nM的siROCK2, Lipo/siROCK2能够显著沉默ROCK2表达。图2-1-2E siNC干扰对ROCK2的表达没有影响。图2-1-3A、A’显示Annexin V-FITC/PI双染流式检测细胞凋亡,结果显示Lipo/siROCK2预处理的凋亡率与METH组相比减少了52.38%,p<0.001,n=3。Lipo/siROCK2组相比Lipo2000或Lipo/siNC处理组相比,可明显的减少PC12细胞的凋亡(F=876.56,*P<0.001,n=3,)。这些结果表明,抑制ROCK2表达可减少METH诱导的PC12细胞的凋亡。图2-1-3B、B’显示TUNEL染色检测凋亡,2.0mM METH处理的PC12细胞TUNEL阳性的细胞数量是对照组的8.9倍,*P<0.01。Lipo/siROCK2预处理2.0mM METH组与2.0mM METH单独处理组相比,TUNEL阳性的细胞数量减少60.2%,P<0.01,接受100nM的siNC预处理组TUNEL阳性的细胞数量却没有明显变化。Lipo/siROCK2预处理组缺乏用箭头所示的典型的凋亡环形核结构。图2-1-4显示,METH+siROCK2组ROS比率与METH组以及METH+siNC组比分别降低了5.3%以及5.1%(*p<0.05,n=3),认为ROCK2抑制可以有效降低METH导致PC12细胞中ROS增加。图2-1-5A显示RhoA的mRNA水平随METH的浓度增加而增加,具有浓度依赖性。2mM的METH处理组RhoA的mRNA比Con组增加了71.84%,**p<0.05,n=3。图2-1-5B显示,尚不能认为siROCK2的使用与METH使用的交互效应对RhoA的mRNA水平的升高有影响(F=42.32,*p>0.05,n=3)。认为METH处理的PC12细胞中,ROCK2的激活可能与RhoA转录水平增加有关。图2-1-5C1Western blot结果显示抑制ROCK2与其逆转细胞凋亡的机制相关。图2-1-5C2显示,METH+siROCK2组p-Akt/Akt比率与METH+Lipo组相比增加了51.0%,p<0.001,n=3。认为ROCK2的抑制可增加METH导致PC12细胞中降低的p-Akt/Akt比率。METH+Lipo组p-Akt/Akt比率与Con组相比降低42.0%,*p<0.01,n=3。认为抑制ROCK2可提高METH处理的PC12细胞中p-Akt/Akt比率。从而促进PC12细胞的存活。图2-1-5C3显示METH+Lipo组cleave PARP/PARP比率与Con组增加了131%,**p<0.001,n=3,认为METH使用导致PC12细胞中PARP的分裂增力。METH+siROCK2组cleave PARP/PARP比率与METH+Lipo组比无统计意义,#p>0.05,n=3。尚不认为siROCK2可减少METH导致增加的cleave PARP/PARP比率。图2-1-5C4显示METH+Lipo组ctive-capspase3与Con组相比增加了200%,**p<0.001,n=3,认为METH使用导致PC12细胞中active-capspase3的增加具有统计学意义。METH+siROCK2组active-capspase3与METH+Lipo组相比无统计学差异,#p>0.05,n=3,尚不认为抑制ROCK2是通过降低caspase3激活这一途径保护神经元。以上说明ROCK2抑制对METH导致PC12细胞的凋亡可能通过通过提高p-Akt/Akt比例,而不是通过抑制METH导致caspase3的激活以及PARP的分裂途径。图2-1-6显示METH处理导致树突解体和突起长度的缩短。然而,PC12形态的变化在METH+Lipo/siROCK2组中得到恢复。图2-1-7显示.抑制ROCK2可部分恢复METH导致原代黑质神经元树突分叉数目的减少以及神经突长度的缩短。图2-1-8显示干扰ROCK2可部分恢复METH导致原代黑质神经元树突棘的密度的降低。图2-1-9A显示干扰ROCK2可部分逆转METH导致原代黑质神经元的轮廓坍塌。图2-1-9B1、B2METH+Saline组的p-MLC/MLC比率与Con/Saline组相比增加54.0%,**p<0.001,n=3,认为METH所导致原代黑质神经元中p-MLC/MLC比率增加具有统计学意义。结果表明干扰ROCK2可降低METH所导致增加的p-MLC/MLC比率,从而增加F-actin的稳定性,有利于增加细胞骨架的稳定性,维持细胞轮廓。第二章第二节图2-2-1免疫荧光以及western blot显示在体模型上ROCK2在METH处理后Str、SN以及PFc区增加明显,而Hip增加不明显。图2-2-2open field检测大鼠自主运动行为结果显示,分组与观察时间的交互效应不显著F=0.16,#P>0.05,认为分组间的差别不随观察时间的不同而改变。采用调整的基于估计边缘值的LSD法进行不同分组间指标的多重指标值的多重比较,Con组、Fasudil组、METH组以及Fasudil组间均无显著性统计学差异P>0.05。METH停药后48h与对照组相比,大鼠自主活动的在各组以及各时间段变化并不具有统计学差异,并未体现出Fasudil对METH损害的大鼠自主行为有保护作用。图2-2-3Western blot显示Fasudil可部分抑制METH导致Str以及SN区中ROCK2的增加,说明抑制ROCK2的METH模型构建成功。图2-2-4中TUNEL染色显示末次METH使用后48h, METH组与Con组比并未在PFc、Str、Hip以及SN区发现TUNEL阳性细胞的增加,不认为此种建模方式导致大鼠神经细胞的凋亡。图2-2-5中尼氏染色显示末次METH使用后48h,MEETH组与Con组比PFc、Str、Hip以及SN区发现尼氏染色阳性的细胞减少,认为此种建模方式导致大鼠神经细胞的损伤。图2-2-6TH染色显示末次METH使用后48h, Str区TH-ir的面积大量减少,Fasudil的使用可以逆转这一改变;SN区TH-ir的神经元数目大量减少,Fasudil的使用可以逆转这一改变。图2-2-7免疫组化结果显示METH可以降低DAT表达以及升高Grp78、Bax、CDK5的表达,而对激活的caspase3改变不明显。Fasudil联合METH使用可部分恢复METH对DAT、Bax、Grp78及CDK5四个指标的改变。提示抑制ROCK2可能通过抑制线粒体损伤、内质网应激、氧化应激对METH损伤上神经系统进行保护。结论:第一章1.体内模型中证实GSTP1表达在METH处理后表达降低。2.GSTP1不在METH处理后的表达降低是由部分由iNOS介导。3.GSTP1对METH导致的氧化应激有抑制作用。4.CDK5可能为GSTP1在METH神经毒性作用机制的下游因子。5.成功构建SN及SN投射区区GSTP1过表达的大鼠模型。第二章1.ROCK2表达的增高具有METH浓度依赖性。2.成功合成高效能的siROCK2,并能成功敲低PC12细胞以及原代中脑神经元中ROCK2的表达。3.抑制ROCK2能部分逆转METH导致PC12细胞的凋亡。4.抑制ROCK2能部分降低METH所导致氧化应激。5.抑制ROCK2通过增加Akt磷酸比例减轻METH导致的凋亡,而并非通过抑制caspase3的激活及PARP的分裂。6.抑制ROCK2能逆转METH导致的PC12细胞形态异常以及原代黑质神经元细胞骨架的不稳定性,维持树突的分支以及长度。其机制可能是通过提高降低p-MLC/MLC去实现的。7.体内实验证实METH处理后ROCK2能在PFc、SN、Str区明显增加,而在Hip区增加不明显。8.在体实验证实抑制ROCK2可保护METH损伤的TH-ir阳性神经末梢以及胞体。9.尼氏体的损伤以及TH染色可能比TUNEL染色检测凋亡更敏感地反应METH导致的神经损伤。10.在体实验提示抑制ROCK2可能通过维持多巴胺的转运水平、抑制线粒体损伤、内质网应激以及氧化应激的途径发挥的保护作用。 还原
1.谷胱甘肽S转移酶1; 周期素依赖性蛋白激酶5; 重组慢病毒; 氧化应激; 凋亡; 硝基化; 甲基苯丙胺; 神经毒性2.Rho相关激酶2; 小RNA干扰; 细胞骨架; 神经毒性;
国家自然科学;
王慧君;
D919.4
康朗生物畅销ELISA试剂盒,良心推荐
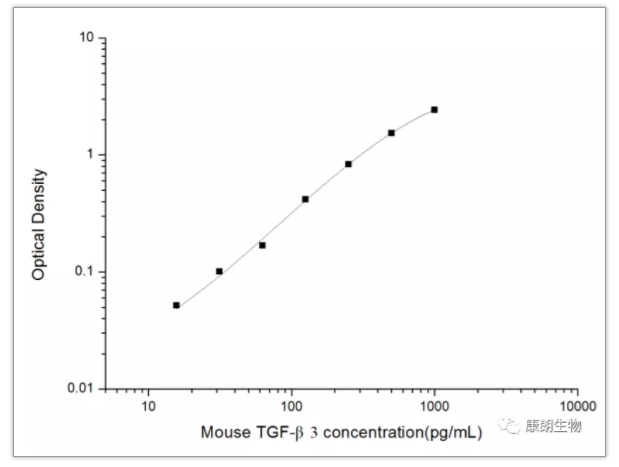
IL-15:多效性细胞因子
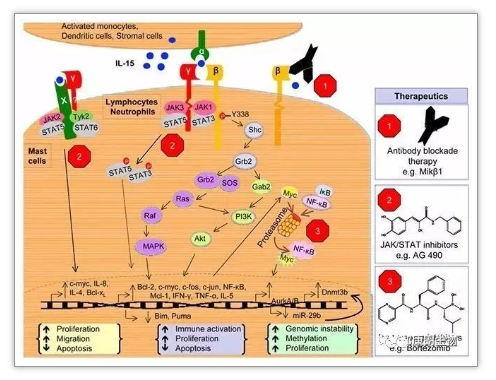
细胞因子在免疫治疗中的应用概述
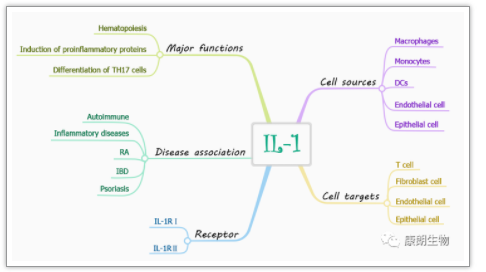
细胞被污染只能扔掉吗?还能抢救一下吗?
相关产品

关注
拨打电话

留言咨询






