1 主题内容与适用范围 本标准样品有土壤和植物材料。本标准适用于校准15N分析的质谱和光谱仪器、检验15N分析方法和各实验室15N分析数据的15N土壤、植物标准样品。 2 15N标准样品的来源及制备 2.1 富集15N土壤样品 取自北京农业大学校园的北京褐土,1983年在微区内施入高丰度15N的液氨,种了一茬水稻。土壤又放置了一年,混匀磨碎,过筛,再充分混匀,装瓶,每瓶30g 。 2.2 富集15N植物样品 水稻茎叶,于田间微区内在后期施入高丰度15N硫铵,待水稻成熟后收割,去掉根和穗子,洗净晾干,切碎,磨成粉,过筛,充分混匀,分装在塑料袋中,每袋2g。 2.3 15N自然丰度植物样品 取自大田的旱稻植株茎叶,经洗净、晾干,切碎,磨成粉末,过筛,充分混匀,分装在塑料袋中,每袋2g。 植物样品和土壤样品封盖后,用钴60γ-射线4.9Mrad照射后,在室温下(10~35℃)保存在暗处。 3 标准样品的细度、均匀度 3.1 细度:植物样品全部通过60目(孔径0.25mm),土壤样品通过100目(孔径0.149mm)筛孔。 3.2 均匀度:随机抽样20袋,经200次测定。富集15N植物样品的丰度为0.691±4.9×10-3,C.V%为0.71;富集15N土壤样品的丰度为0.441±2.9×10-3,C.V%为0.66。结果表明,样品是混合均匀的。 4 标准样品的标准值 协作组对本标准样品进行了多次反复,长期的测定,结果如下: 样品名称 15N原子% N% 15N自然丰度植物样品 0.366±0.001 1.026 富集15N植物样品 0.688±0.003 0.745 富集15N土壤样品 0.441±0.003 0.077 注:N%的参比值,供参考。 5 标准样品的保存和使用方法 5.1 避光、干燥保存,温度不超过30℃。 5.2 使用前先用牛角勺搅动,使再度混匀。 5.3 一般分析用量:土壤至少称取1g,植物样品至少0.2g。

[size=16px] 植物根系分析仪的检测结果准确性取决于多个因素,包括仪器的质量和性能、操作的正确性、样本的准备和处理等。以下是一些影响植物根系分析仪检测结果准确性的关键因素: 仪器质量和性能: 使用高质量的根系分析仪通常会提高结果的准确性。精密仪器通常具有更高的分辨率和稳定性,能够更精确地测量根系的参数。 操作的正确性: 操作人员需要按照仪器的操作手册和相关方法正确地操作仪器。错误的操作可能导致结果的偏差。 样本准备和处理: 样本的准备和处理对于根系分析的准确性至关重要。根系样本应该被适当地清洗、固定和处理,以避免任何外部因素的干扰。 数据分析和解释: 数据的分析和解释也是确保准确结果的重要步骤。使用适当的分析方法和软件来处理和解释数据是关键。 环境因素: 根系分析的环境因素,如温度、湿度和光照条件,也可能影响结果的准确性。这些因素需要在分析中加以考虑。 根系生长阶段: 不同生长阶段的植物根系可能具有不同的形态和特性。因此,在分析时需要考虑植物的生长阶段。 根系分析仪的校准: 定期校准根系分析仪以确保其性能和测量的准确性是重要的。校准可以帮助纠正仪器的误差。 总之,为了确保云唐植物根系分析仪的检测结果准确,需要注意上述因素,并严格按照操作规程执行。此外,可以通过与其他方法或仪器的比较来验证结果的准确性,以确保所得数据的可靠性。[img=,690,690]https://ng1.17img.cn/bbsfiles/images/2023/09/202309131026366807_8752_6098850_3.jpg!w690x690.jpg[/img][/size]

[size=16px] 手持式植物养分速测仪如何检测植物叶面温度 手持式植物养分速测仪通常不用于测量叶面温度,而是用于测量植物的营养元素含量、叶绿素含量等参数。要测量叶面温度,通常需要使用红外热像仪或红外温度计等专门的仪器。以下是如何使用红外热像仪来测量植物叶面温度的一般步骤: 准备手持式植物养分速测仪: 打开手持式植物养分速测仪,并确保它已经达到稳定的工作状态。 根据仪器的使用说明,进行必要的校准和设置。 准备测量环境: 在测量之前,确保测量环境没有明显的干扰因素,如直射阳光、风、或其他热源。 将手持式植物养分速测仪对准要测量的植物叶面区域。 进行测量: 按下手持式植物养分速测仪上的触发按钮来拍摄或记录叶面的红外热图像。 等待仪器处理图像数据,以获取叶面温度信息。 手持式植物养分速测仪可以直接显示叶面温度,而其他仪器可能需要将数据传输到计算机或移动设备上进行分析。 分析结果: 分析所获得的红外热图像,查看叶面温度的分布情况。 记录或分析所需的温度数据,以了解植物的温度状况。 云唐手持式植物养分速测仪能够测量物体表面的温度,因此可以用于监测植物叶面的温度分布,以帮助农业和植物研究人员更好地理解植物的生长和健康状态。要获得准确的叶面温度数据,确保仪器的使用和环境设置是适当的,并根据仪器的说明进行操作。[img=,690,690]https://ng1.17img.cn/bbsfiles/images/2023/09/202309181128595765_5081_6098850_3.jpg!w690x690.jpg[/img][/size]
[font=-apple-system, BlinkMacSystemFont, &][color=#05073b][size=16px] 植物呼吸测定仪的误差范围是多少,植物呼吸测定仪(如KZJ-04型号)的误差范围在参考文章中并未直接给出具体的数值。然而,从一般测试仪器的通用性和准确性角度来看,误差范围可能会受到多种因素的影响,如仪器的设计、校准、操作条件等。 以下是对植物呼吸测定仪误差范围可能涉及的一些方面的归纳和解释: 仪器设计和技术指标: 植物呼吸测定仪(如KZJ-04型号)采用非扩散式红外CO?分析来测量CO?浓度,这是影响呼吸速率测定的关键因素之一。红外CO?分析器的精度和稳定性将直接影响呼吸速率的测量准确性。 技术指标中提到的测量范围(如0-2000ppm/0-1500ppm可选)可能暗示了仪器在此范围内的测量能力,但具体的误差范围需要参照仪器的校准证书或制造商提供的技术规格。 校准和验证: 植物呼吸测定仪在使用前和使用过程中需要进行定期的校准和验证,以确保其测量结果的准确性。校准通常涉及使用已知浓度的气体样品来检验仪器的响应。 校准过程中可能会提供仪器的误差范围或准确度信息,这些信息是评估仪器测量可靠性的重要依据。 操作条件和样品特性: 植物呼吸速率的测量受到多种操作条件(如温度、湿度、光照等)和样品特性(如植物种类、生长状态、叶片大小等)的影响。这些因素可能导致测量结果的波动和误差。 因此,在使用植物呼吸测定仪时,需要确保操作条件的稳定性和一致性,并尽可能减少样品特性的差异对测量结果的影响。 总结: 由于缺乏具体的误差范围数值,我们无法直接给出植物呼吸测定仪(如KZJ-04型号)的误差范围。然而,通过了解仪器的设计原理、技术指标、校准和验证过程以及操作条件和样品特性的影响,我们可以对仪器的测量准确性有一个大致的评估。 在实际应用中,建议参考制造商提供的技术规格和校准证书,并结合实际使用经验来评估植物呼吸测定仪的测量误差范围。[img=,690,690]https://ng1.17img.cn/bbsfiles/images/2024/05/202405291109183963_977_6098850_3.jpg!w690x690.jpg[/img][/size][/color][/font]
各相关机构和人员: 中国合格评定国家认可委员会(CNAS)组织制定了CNAS-CL01-A014:202X《检测和校准实验室能力认可准则在植物检疫领域的应用说明》。目前已完成文件征求意见稿,现于网上公示征求意见。 请相关机构和人员对该文件有修改建议或意见,请填写附件中的意见征询表,并于2024年3月18日前反馈至CNAS秘书处。 联系人:富宏坤 联系电话:010-67105451 Email:fuhk@cnas.org.cn
[size=16px] 叶面积测量仪是用于测量植物叶片表面积的工具。它可以通过不同的方法来估算叶片的表面积,其中一些常见的方法包括: 直接测量法: 这种方法涉及将叶片放在一块已知面积的测量板上,然后使用划分网格或数字图像处理来测量叶片的轮廓。然后可以使用这些测量值来计算叶片的总表面积。 扫描法: 这种方法使用数码扫描仪或图像扫描仪来扫描叶片的图像。扫描后,使用图像处理软件测量叶片的轮廓,并计算出叶片的表面积。 影像分析法: 使用数字相机或移动设备拍摄叶片的图像,然后使用专业的图像分析软件来处理图像,提取叶片的轮廓并测量表面积。 数学模型法: 通过测量叶片的长度、宽度和其他几何特征,然后应用数学模型(如椭圆形、矩形等)来估算叶片的表面积。 叶片分段法: 对于大型或不规则形状的叶片,可以将其分成几个较小的部分,测量每个部分的面积,然后将这些面积相加以得到总表面积。 无论使用哪种方法,都需要确保测量精确度和可靠性。在使用叶面积测量仪进行测量时,云唐建议遵循制造商提供的操作说明,并根据需要进行校准,以确保获得准确的叶面积数据。另外,不同类型的植物可能需要针对其特定叶片形状和大小的方法进行微调。[/size]
[font=&][size=18px]一、植物全氮测定[/size][/font] [font=&][size=18px](一)H2SO4-H2O2消煮法[/size][/font] [font=&][size=18px]1、适用范围[/size][/font] [font=&][size=18px]本方法不包括硝态氮的植物全氮测定,适合于含硝态氮低的植物样品的测定。[/size][/font] [font=&][size=18px]2、方法提要[/size][/font] [font=&][size=18px]植物中的氮、磷大多数以有机态存在,钾以离子态存在。样品经浓H2SO4和氧化剂H2O2消煮,有机物被氧化分解,有机氮和磷转化成铵盐和磷酸盐,钾也全部释出。消煮液经定容后,可用于氮、磷、钾的定量。采用H2O2为加速消煮的氧化剂,不仅操作手续简单快速,对氮、磷、钾的定量没有干扰,而且具有能满足一般生产和科研工作所要求的准确度。但要注意遵照操作规程的要求操作,防止有机氮被氧化成N2气或氮的氧化物而损失。[/size][/font] [font=&][size=18px]3、试剂[/size][/font] [font=&][size=18px](1)硫酸(化学纯,比重1.84) [/size][/font] [font=&][size=18px](2)30% H2O2(分析纯)。[/size][/font] [font=&][size=18px]4、主要仪器设备。消煮炉,定氮蒸馏器。[/size][/font] [font=&][size=18px]5、操作步骤[/size][/font] [font=&][size=18px]称取植物样品(0.5mm)0.3~0.5g(称准至0.0002g)装入100ml开氏瓶或消煮管的底部,加浓H2SO45ml,摇匀(最好放置过夜),在电炉或消煮炉上先小火加热,待H2SO4发白烟后再升高温度,当溶液呈均匀的棕黑色时取下。稍冷后加班10滴H2O2(3),再加热至微沸,消煮约7~10min,稍冷后重复加H2O2,,再消煮。如此重复数次,每次添加的H2O2应逐次减少, 消煮至溶液呈无色或清亮后,再加热10min,除去剩余的H2O2。取下冷却后,用水将消煮液无损地转移入100ml容量瓶中,冷却至室温后定容(V1)。用无磷钾的干滤纸过滤,或放置澄清后吸取清液测定氮、磷、钾。每批消煮的同时,进行空白试验,以校正试剂和方法的误差。[/size][/font] [font=&][size=18px]6、注释[/size][/font] [font=&][size=18px](1)所用的H2O2应不含氮和磷。H2O2在保存中可能自动分解,加热和光照能促使其分解,故应保存于阴凉处。在H2O2中加入少量H2SO4酸化,可防止H2O2分解。[/size][/font] [font=&][size=18px](2)称样量决定于NPK含量,健状茎叶称0.5g,种子0.3g,老熟茎叶可称1g,若新鲜茎叶样,可按干样的5倍称样。称样量大时,可适当增加浓H2SO4用量。[/size][/font] [font=&][size=18px](3)加H2O2时应直接滴入瓶底液中,如滴在瓶劲内壁上,将不起氧化作用,若遗留下来还会影响磷的显色。[/size][/font] [font=&][size=18px](二)水杨酸-锌粉还原- H2SO4-加速剂消煮法[/size][/font] [font=&][size=18px]1、适用范围[/size][/font] [font=&][size=18px]包括销态氮的植物全氮测定,适合于硝态氮含量较高的植物样品的测定。[/size][/font] [font=&][size=18px]2、方法原理[/size][/font] [font=&][size=18px]样品中的硝态氮在室温下与硫酸介质中的水杨酸作用,生成硝基水杨酸,再用硫代硫酸钠及锌粉使硝基水杨酸还原为氨基水杨酸.然后按H2SO4-加速剂消煮法进行消煮法进行消煮样品,使样品中全部氮转化为铵盐。[/size][/font] [font=&][size=18px]3、试剂[/size][/font] [font=&][size=18px](1)固体Na2S2O3 [/size][/font] [font=&][size=18px](2)还原锌粉(AR) [/size][/font] [font=&][size=18px](3)水杨酸-硫酸:30g水杨酸溶于1L浓硫酸中。也可以该用含苯酚的浓硫酸:40g苯酚溶于1L浓硫酸中。[/size][/font] [font=&][size=18px]4、仪器设备。同上。[/size][/font] [font=&][size=18px]5、操作步骤[/size][/font] [font=&][size=18px]称取磨细烘干样品(过0.25mm筛)0.1000~0.2000g或新鲜茎叶样品1.000~2.000g,置于100ml开氏瓶或消煮管中,先用水湿润内样品(烘干样),然后加水杨酸-硫酸10ml,摇匀后室温放置30min,加入Na2S2O3约1.5g,锌粉0.4g和水10ml,放置10 min,待还原反应完成后,加入混合加速剂2g,按土壤全氮测定方法进行消煮, 消煮完毕,取下冷却后,用水将消煮液无损地转移入100ml容量瓶中,冷却至室温后定容(V1)。用于滤纸过滤,或放置澄清后吸取清液测定氮。每批消煮的同时,进行空白试验,以校正试剂和方法的误差。[/size][/font] [font=&][size=18px](三)消煮液中铵的定量(凯氏法)[/size][/font] [font=&][size=18px]1、适用范围。适合于各种植物样品消煮液中氮的定量。[/size][/font] [font=&][size=18px]2、方法原理[/size][/font] [font=&][size=18px]植物样品经开氏消煮、定容后,吸取部分消煮液碱化,使铵盐转变成氨,经蒸馏,用H3BO3吸收,硼酸中吸收的氨可直接用标准酸滴定,以甲基红-溴甲酚绿混合指示剂指标终点。[/size][/font] [font=&][size=18px]3、试剂[/size][/font] [font=&][size=18px](1)400g/L NaOH溶液。[/size][/font] [font=&][size=18px](2)20g/L H3BO3-指示剂溶液。[/size][/font] [font=&][size=18px](3)酸标准溶液[c(HCL或1/2H2SO4)=0.01mol/L]。[/size][/font] [font=&][size=18px]4、仪器设备。蒸馏装置或半自动蒸馏仪。[/size][/font] [font=&][size=18px]5、蒸馏[/size][/font] [font=&][size=18px]检查蒸馏装置是否漏气和管道是否洁净后,吸取定容后的消煮液5.00~10.00mL (V2,含NH4-N约1mg),注入半微量蒸馏器的内室。另取150ml三角瓶,内加5 ml 2% H3BO3指示剂溶液(若为包括硝态氮的待测液,应加约6 mL的400g/L NaOH溶液),通过蒸气蒸馏(注意开放冷凝水,勿使馏出液温度超过40℃)。待馏出液体积约达50~60ml时,停止蒸馏,用少量已调节至pH4.5的水冲洗冷凝管末端。用酸标准溶液滴定馏出液至由蓝绿色突变为紫红色(终点的颜色应和空白测定的滴定终点相同)。与此同时进行空白测定的蒸馏、滴定、以校正试剂和滴定误差。[/size][/font] [font=&][size=18px]6、结果计算[/size][/font] [font=&][size=18px]ω(N), %=c(V-V0)×0.014×D×100/m [/size][/font] [font=&][size=18px]式中: ω(N)——植物全氮的质量分数,% [/size][/font] [font=&][size=18px]c——酸标准溶液的浓度,mol/L [/size][/font] [font=&][size=18px]V——滴定试样所用的酸标准液体积,ml [/size][/font] [font=&][size=18px]V0——滴定空白所用的酸标准液, ml [/size][/font] [font=&][size=18px]0.014——N的摩尔质量,kg/mol [/size][/font] [font=&][size=18px]D——分取倍数(即消煮液定容体积V1/吸取测定的体积V2)。[/size][/font] [font=&][size=18px]二、植物全磷的测定[/size][/font] [font=&][size=18px](一) 钒钼黄吸光光度法[/size][/font] [font=&][size=18px]1、适用范围。适合于含磷量较高的植物样品的测定(如籽粒样品)。[/size][/font] [font=&][size=18px]2、方法原理[/size][/font] [font=&][size=18px]植物样品经浓H2SO4消煮使各种形态的磷转变成磷酸盐。待测液中的正磷酸与偏钒酸和钼酸能生成黄色的三元杂多酸,其吸光度与磷浓度成正比,可在波长400~490nm处用吸光光度法测定。磷浓度较高时选用较长的波长,较低时选用较短波长。[/size][/font] [font=&][size=18px]此法的优点是操作简便,可在室温下显色,黄色稳定,在HNO3、HClO4和H2SO4等介质中都适用,对酸度和显色剂浓度的要求也不十分严格,干扰物少,在可见光范围内灵敏度较低,适测范围广(约为1~20mg/L P),故广泛应用于含磷较高而且变幅较大的植物和肥料样品中磷的测定。[/size][/font] [font=&][size=18px]3、试剂[/size][/font] [font=&][size=18px](1)钒钼酸铵溶液:25.0g钼酸铵[(NH4)6Mo7O24H2O,分析纯]溶于400mL水中,必要时可适当加热,但温度不得超过60℃。另将1.25g偏钒酸铵(NH4VO3,分析纯)溶于300mL沸水中,冷却后加入250mL浓HNO3(分析纯)。将钼酸铵溶液缓缓注入钒酸铵(溶液中,不断搅匀,最后加水稀释至1L,贮于棕色瓶中。[/size][/font] [font=&][size=18px](2)NaOH溶液(c=6mol/L):24gNaOH溶于水, 稀释至100ml。[/size][/font] [font=&][size=18px](3)二硝基酚指示剂(ρ=2g/L):0.2g2,6-二硝基酚或2,4-二硝基酚溶于100ml水中。[/size][/font] [font=&][size=18px](4)磷标准溶液ρ[(P)=50mg/L]:0.2195g(干燥的KH2PO4(分析纯)溶于水,加入5ml浓HNO3,于1L容器瓶中定容。[/size][/font] [font=&][size=18px]4、主要仪器设备。分光光度计。[/size][/font] [font=&][size=18px]5、分析步骤[/size][/font] [font=&][size=18px]准确吸取定容,过滤或澄清后的消煮液5~20ml(V2,含P0.05~0.75mg)放入50ml容量瓶中,加2滴二硝基酚指示剂,滴加6mol/LNaOH中和至刚呈黄色,加入10.00ml钒钼酸铵试剂,用水定容(V3)。15min后,用1cm光径的比色槽在波长440nm处进行测定,以空白溶液(空白溶液消煮液按上述步骤显色),调节仪器零点。[/size][/font] [font=&][size=18px]校准曲线或直线回归方程:准确吸取50mg/L P标准液0, 1, 2.5, 7.5, 10, 15ml分别放入50mL容量瓶中,按上述步骤显色,即得0, 1.0, 2.5 , 5.0, 7.5, 10, 15 ml P的标准系列溶液,与待测液一起进行测定,读取吸光度,然后绘制校准曲线或求直线回归方程。[/size][/font] [font=&][size=18px]6、结果计算[/size][/font] [font=&][size=18px]ρ(P)×V3×(V1/V2)×10-4[/size][/font] [font=&][size=18px]ω(P)=[/size][/font] [font=&][size=18px]m[/size][/font] [font=&][size=18px]式中: ω(P) ——植物磷的质量分数,% [/size][/font] [font=&][size=18px]ρ(P) ——从校准曲线或回归方程求得的显色液中磷的质量浓度, mg/L [/size][/font] [font=&][size=18px]V1——消煮液定容体积, ml [/size][/font] [font=&][size=18px]V2——吸取测定的消煮液体积, ml [/size][/font] [font=&][size=18px]V3——显色液体积, ml [/size][/font] [font=&][size=18px]m——称样量,g [/size][/font] [font=&][size=18px]10-4——将mg/L浓度单位换算为百分含量的换算因数。[/size][/font] [font=&][size=18px]7、注释[/size][/font] [font=&][size=18px](1)显色液中ρ(P)=1~5 mg/L时,测定波长420nm 5~20mg/L用490nm。待测液中Fe3+浓度高应选用450nm,以清除Fe3+干扰。校准曲线也应用同样波长测定绘制。[/size][/font] [font=&][size=18px](2)一般室温下,温度对显色影响不大,但室温太低(如15℃)时,需显色30min。稳定时间可达24h。[/size][/font] [font=&][size=18px](3)如试液为HCl,HClO4介质,显色剂应用HCl配制 试液为H2SO4介质, 显色剂也用H2SO4配制。显色液酸的适宜浓度范围为0.2~1.6 mol/L,最好是0.5~1.0 mol/L。酸度高显色慢且不完全,甚至不显色 低于0.2 mol/L易产生沉淀物, 干扰测定。钼酸盐在显色液中的终浓度适宜范围为1.6×10-3~10-2mol/L, 钒酸盐为8×10-5~2.2×10-3 mol/L。[/size][/font] [font=&][size=18px]4、此法干扰离子少。主要干扰离子是铁,当显色液中Fe3+浓度超过0.1%时,它的黄色有干扰,可用扣除空白消除。[/size][/font] [font=&][size=18px](二)钼锑抗吸光光度法[/size][/font] [font=&][size=18px]1、适用范围[/size][/font] [font=&][size=18px]适合于含磷量较低的植物样品的测定(如茎秆样品等)。[/size][/font] [font=&][size=18px]2、方法提要[/size][/font] [font=&][size=18px]植物样品经浓H2SO4消煮使各种形态的磷转变成磷酸盐。在一定酸度下,待测液中的正磷酸与钼酸铵和酒石酸锑钾生成一种三元杂多酸,后者在室温下能迅速被抗坏血酸还原为蓝色络合物,可用吸光光度法测定。[/size][/font] [font=&][size=18px]3、试剂[/size][/font] [font=&][size=18px](1)6mol/L NaOH溶液[/size][/font] [font=&][size=18px](2)0.2%二硝基酚指示剂[/size][/font] [font=&][size=18px](3)2mol/L(1/2 H2SO4)硫酸溶液:5.6mL浓H2SO4加水至100mL。[/size][/font] [font=&][size=18px](4)钼锑贮存液: 浓H2SO4(分析纯)126 ml缓慢地注入约400 ml水中,搅拌,冷却。10.0g钼酸铵(分析纯)溶解于约60℃的300ml水中,冷却。然后将H2SO4溶液缓缓倒入钼酸铵溶液中,再加入100 ml0.5%酒石酸锑钾(KSbOC4O61/2H2O, 分析纯) 溶液,最后用水稀释至1L,避光贮存。此贮存液含钼酸铵为1%,酸浓度为c(1/2 H2SO4)=4.5 mol/L[/size][/font] [font=&][size=18px](5)钼锑抗显色剂:1.50g抗坏血酸(C6H8O6,左旋,旋光度+21~+22, 分析纯) 溶于100ml钼锑贮存液中,此液须随配随用,有效期一天,冰箱中存放,可用3~5天。[/size][/font] [font=&][size=18px](6)磷标准工作液[ρ(P)=5 mg/L]:吸取100mg/L P标准贮存液稀释20倍,即为5 mg/L P标准工作溶液,此溶液不宜久存。[/size][/font] [font=&][size=18px]4、主要仪器设备。同上[/size][/font] [font=&][size=18px]5、分析步骤[/size][/font] [font=&][size=18px]吸取定容过滤或澄清后的消煮液2.00~5.00ml(V2,含P5~30ug)于50ml容量瓶中, 用水稀释至约30ml,加1~2滴二硝基酚指示剂,滴加6mol/L NaOH溶液中和至刚呈黄色,再加入1滴2mol/L(1/2 H2SO4)溶液,使溶液的黄色刚刚褪去,然后加入钼锑抗显色剂5.00ml,摇匀,用水定容(V3)。在室温高于15℃的条件下放置30min后,用1cm光径比色槽在波长700nm处测定吸光度,以空白溶液为参比调节仪器零点。[/size][/font] [font=&][size=18px]校准曲线或直线回归方程: 准确吸取ρ(P)= 5mg/L标准工作溶液0, 1, 2, 4, 6, 8 ml,分别放入50mL容量瓶中,加水至30ml,同上步骤显色并定容, 即得0,按0.1, 0.2, 0.4, 0.6, 0.8 mg/L P标准系列溶液, 与待测液同时测定,读取吸光度,然后绘制校准曲线或直线回归方程。[/size][/font] [font=&][size=18px]6、结果计算:同1。[/size][/font] [font=&][size=18px]7、注释[/size][/font] [font=&][size=18px]根据分光光度计性能,可选用650~890nm波长处测定,880~890nm处灵敏度高[/size][/font] [font=&][size=18px]三、植物全钾的测定—火焰光度法[/size][/font] [font=&][size=18px](一)适用范围。适合于植物样品消煮液中钾含量的测定。[/size][/font] [font=&][size=18px](二)方法提要[/size][/font] [font=&][size=18px]植物样品经消煮或浸提,并经稀释后,待测液中的K可用火焰光度法测定。[/size][/font] [font=&][size=18px](三)试剂[/size][/font] [font=&][size=18px]K标准溶液[ρ(K)= 100mg/L] :0.1907gKCl(分析纯),在105~110℃干燥2h)溶于水,于1L容量瓶中定容,存于塑料瓶中。[/size][/font] [font=&][size=18px](四)主要仪器设备。火焰光度计。[/size][/font] [font=&][size=18px](五)分析步骤[/size][/font] [font=&][size=18px]吸取定容后的消煮液5.00~10.00ml(V2)放入50mL容量瓶中,用水定容(V1),直接在火焰光度计上测定,读取检流计读数。[/size][/font] [font=&][size=18px]校准曲线或直线回归方程 准确吸取100mg/L K标准溶液0, 1, 2.5, 10, 20 ml, 分别放入50mL容量瓶中,加水定容的空白消煮液5或10ml(使标准溶液中的离子成分和待测[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相[/color][/url]近),加水定容。即得0, 2, 5, 10, 20, 40 mg/L K标准系列溶液。以浓度最高的标准溶液定火焰光度计检流计的满度(一般只定到90),然后从稀到浓依次进行测定,记录检流计读数,以检流计读数为纵坐标,钾浓度为横坐标绘制校准曲线或求直线回归方程。[/size][/font] [font=&][size=18px](六)结果计算[/size][/font] [font=&][size=18px]ρ(K)×V3×(V1/V2)×10-4[/size][/font] [font=&][size=18px]ω(K)=[/size][/font] [font=&][size=18px]m[/size][/font] [font=&][size=18px]式中: ω(K) ——植物钾的质量分数,% [/size][/font] [font=&][size=18px]ρ(K) ——从校准曲线或回归方程求得的测读液中K的浓度, mg/L [/size][/font] [font=&][size=18px]V1——消煮液定容体积, ml [/size][/font] [font=&][size=18px]V2——吸取体积, ml [/size][/font] [font=&][size=18px]V3——测读液定容体积, ml [/size][/font] [font=&][size=18px]m——干样质量,g [/size][/font] [font=&][size=18px]10-4——将mg/L浓度单位换算为百分含量的换算因数[/size][/font]
由于工矿企业的发展,农业化肥的过量使用,污水灌溉等,中国乃至世界的土壤重金属污染越来越严重。植物修复技术是目前重金属污染治理的研究热点,它具有治理效果的永久性、治理过程的原位性、治理成本的低廉性、环境美学的兼容性、后期处理的简易性等优点。这个技术成功的关键在于寻找超富集植物。虽然目前全世界已发现400多种超富集植物,但是大多数超富集植物都有生物量小,生长缓慢,弱抵抗力,种子少,缺乏与当地植物竞争的能力等缺点,所以能够真正应用于植物修复技术的超富集植物并不多。因此采用更有效的方法来筛选更多超富集植物是非常必要的。 华南植物园土壤生态与生态工程研究组博士研究生张杏锋在导师夏汉平研究员的指导下,首次提出了用土壤种子库-重金属浓度梯度法来筛选重金属超富集植物并成功找到一种Cd的超富集植物—少花龙葵(Solanum photeinocarpum)。该方法是指利用土壤种子库筛选对重金属具有超富集特性的植物,然后通过重金属浓度梯度实验对其超富集特性进行验证。结果发现,当土壤Cd 浓度为60mg/kg时,少花龙葵的生长未受影响,根部Cd含量高达473mg/kg,茎、叶和地上部Cd含量分别达215、251和230mg/kg。在两个浓度梯度实验中少花龙葵地上部Cd含量均超过Cd超富集植物的临界含量标准(100mg/kg),具有Cd超富集植物的基本特征,是Cd的超富集植物。
日本植物油中酸价的标准酸价的国标和行标又是怎样的?急!!急
本报讯一项关于促进野生药用植物和芳香植物可持续管理与贸易的新标准,日前在德国纽伦堡颁布。该标准的出台将有助于保证用于医药和化妆品的植物不被过度利用。 此项新标准由世界自然保护联盟物种保护委员会药用植物专家组编制。据该专家组报告,全球大约有1.5万种药用和芳香植物处于濒危状态,占全球所有药用和芳香植物的21%。 全球每年的药用植物和芳香植物贸易量为40多万吨,其中超过80%的植物采自野外,涉及7万种植物,由于过度采集或其栖息地遭到破坏,其中许多濒临灭绝。例如,印度有319种药用植物被世界自然保护联盟列为受威胁物种。 在东欧,过度采集野生药用植物“雉眼”或所谓的“春天的阿多尼斯”作为强心剂的原料,导致这种植物在整个分布区数量急剧下降。目前在许多国家,这种植物已被禁止采集。 在美国,大量野生美国人参和白毛茛被采挖。虽然美国目前已能人工生产美国人参,但仍然存在采挖野生人参现象。现在,这些植物的贸易已有严格规范,两者均被列入濒危物种国际贸易公约附录Ⅱ。列入濒危物种国际贸易公约附录Ⅱ的植物,必须通过审批才能进行贸易。每年大约有90%的美国人参出口东亚。而美国所进口的各种药用植物达成千上万吨,支撑着其价值30亿美元的市场。 世界自然保护联盟物种保护委员会药用植物专家组在与植物专家和药用植物企业进行了广泛的研讨后,起草了该国际野生草药和芳香植物采挖标准(ISSC-MAP)。
由于近期要测定几项动植物油项目,但苦于没有相关检测标准,如果哪位有以下相关检测方法(相关国标也可),请联系我,不胜感激!ISO 660:1996 动植物脂肪和油――酸价和酸度的测定ISO 8534:1996 动植物脂肪和油――水分含量的测定――卡尔费休法ISO 661:1989 动植物脂肪和油――试样的制备ISO 663:1992 动植物脂肪和油――不溶性杂质含量的测定
想购买一些土壤、植物标准物质,请求推荐几家。谢谢!
诸位坛友好,我是新入行的一位质量人员,现在我公司准备做一款植物饮料,需要进行质量控制,其中微生物这一块必然是重中之重,我有问题不解:植物饮料的微生物标准是依据《GB-T 23784-2009 食品微生物指标制定和应用的原则》,但是具体要检测微生物是参考GB4789系列,植物饮料如果参考GB/T 4789.21的话,其中并不包括其他饮料(植物饮料在1996年的饮料分类中属于其他饮料,而推荐标准中也没有其他饮料)那么是不是意味着我要检测GB4789中所有提到的微生物种类?
作植物灰分的分析时,用硝酸,高氯酸分解样品 用盐酸(1+50)把样品稀释到合适范围内。同时取适量的水用与样品相同的方法制成空白溶液。用空白结果去校正测定值。校准曲线:取5至10ml的Zn标准溶液(10ppm)以逐渐增加的体积到100ml容量瓶中,然后加与样品相同量的酸,用水定容到刻度。不知道它是如何用“空白结果去校正测定值”? 具体步骤如何操作(比如测得的空白结果为5ppm,而样品测定结果为20ppm,则实际结果为多少? )?另外它那个校准曲线也不理解是怎么回事标准曲线是基准物质用去离子稀释还是用盐酸稀释好?比如测得的空白结果为5ppm,而样品测定结果为20ppm,则实际结果为多少?

植物激素为何在生产蔬果的大棚和大田中大行其道http://ng1.17img.cn/bbsfiles/images/2011/06/201106150855_299880_2185349_3.jpg在正常使用情况下, 植物激素进入蔬菜体内会随着新陈代谢的进行逐渐降解,药效慢慢消失,在蔬菜体内的残留量很低,即使有微量的残留,在煎炒烹炸的过程中也会被破坏不知从什么时候开始,市场上“顶花带刺”的黄瓜越来越多,而黄瓜顶端的小黄花和瓜体上的疣刺似乎已经成为判断黄瓜鲜嫩于否的标志。不久前有媒体揭开了其中的秘密,原来这些黄瓜是通过涂抹植物激素来“扮嫩”的。一时间,植物激素又被推进了健康问题舆论的漩涡。植物激素并非什么新的化工产品,将它们应用在蔬果生产也不是什么新鲜事,在北方的冬天,能吃上黄澄澄的香蕉和品相完美的西红柿,这里面都有植物激素的功劳。准确地说,目前生产中使用的是与植物激素效应类似的化学物质(如2,4-D,萘乙酸等)。有人提出将其中植物自身分泌的称为植物激素,将人工合成的称为植物生长调节剂。不过在大多数情况下,还是将这些成分都称为植物激素。只是在这个越来越关注健康的时代,大家更关心恐怕不是它们的名字,而是这些非天然的化学品对我们的健康究竟有没有影响。
各位老师,GB5009.262-2016测定植物油中溶剂残留,基体植物油怎么选,找个压榨油超声高温可以吗?这个标准曲线是怎么建的,需要峰面积加和,但是有截距,未检出样品计算得到负值
植物生长调节剂是在生物科学理论指导下,采用化学合成或微生物发酵的方法制取的,具有特定生理调节功能的简单有机化合物,可用于调节植物的生长和发育且低毒或微毒,在增强植物的抗逆性以及在促根、保果、保鲜、提质和增产等方面,都具有十分明显的效果,在国内外的农业生产领域已经得到了广泛的应用。目前,全球植物生长调节剂的销售额约15亿美元,占农药总销售额的5%左右,并以每年10%速度增长;其中,美国EPA批准登记的植物生长调节剂成分已有20多种、产品达200多个。我国生产使用植物生长调节剂也有30多年的历史,目前已登记的植物生长调节剂产品有587个,涉及有效成分近40个,正处于成长发展的阶段。 今年以来,国内连续出现有关“乙烯利催熟香蕉危害人体健康”的不实报道以及“膨大剂导致西瓜爆炸”和“激素黄瓜”的报道,使植物生长调节剂的应用备受质疑。不仅在广大消费者中造成恐慌心理,也给农产品供应、农民增收乃至植物生长调节剂产业的发展,带来了不利的影响。上述现象的产生,有着错综复杂的原因,当前主要存在以下4个方面的问题:
强力机校准的时候无法清零是怎么回事?上面的数字一直在跳,而且无法启动,就算启动了 也就一秒就停了

针对织物撕裂仪(如下图片所示)力值校准,按照JJF(纺织)049-2012 摆锤式织物撕裂仪校准规范 条款5.2.4.2示值误差为±1%FS.是否可理解只要最大量程对应的校准点符合±1%即可?以下校准报告是否符合JJF的要求?[img=,690,479]https://ng1.17img.cn/bbsfiles/images/2022/12/202212271900233673_5943_2902363_3.jpg!w690x479.jpg[/img][img=,690,410]https://ng1.17img.cn/bbsfiles/images/2022/12/202212271854122656_2878_2902363_3.jpg!w690x410.jpg[/img]
急求!做大气中重金属时,有时会附带有树叶等植物样品,但是没有资质认证不能出CMA报告啊!参照食品的方法又说只有卫生部门才能做!请问大家知道有其他关于测定植物样品重金属的标准方法或权威书刊吗?可以做计量认证的。无尽感激!!
根据CANS-TRL-006:2018轻纺实验室测量设备的计量溯源或核查工作指南,织物透气量仪校准参数要校准[color=#ff0000]压力[/color]和[color=#ff0000]流量[/color]两个参数。大家知道有哪里的计量机构可以校准这两个参数?目前深圳计量院仅能校压力。
植物药:来自于传统草药中的药品Shaw T Chen,Jinhui Dou,Robert Temple,Rajiv Agarwal,Kuei-Meng Wu,Susan Walker 尽管植物新药对制药行业与FDA提出许多新的挑战,但是,第一个植物处方药的批准表明他们可以成功地面对这些挑战。2006年10月31日,美国食品药品管理局(Food and Drug Administration,FDA)批准了Veregen (sinecatechins)的新药上市申请(new drug application,NDA),其为治疗外生殖器和肛周疣的外用药。与大多数由单一化合物组成的小分子药物不同,Veregen(绿茶树叶提取物)含有已知的并且可能是活性化合物的混合物。自2004年7月FDA发布《植物药新药指南》[1]以来,Veregen是第一个得到FDA批准的植物新处方药。Veregen得到批准表明天然复杂混合物能够被开发为符合目前FDA对于质量控制标准与临床试验要求的新药。近几年,人们对传统方法生产的草药或植物药的关注程度逐步地提高。1982-2007年,350多个新药临床研究(IND)申请与新药临床前研究咨询(pre-IND)申请递交到FDA。但是,对于如此复杂的产品能否符合目前研究标准的可行性仍有疑问,植物药新药研制的整个进展也较为缓慢。本文描述了当前美国植物药新药研制所处的监管环境、监管经验总结、以及涉及的科学监管问题。我们希望对第一个批准的植物药新药的介绍将促进对潜在有用的植物药产品开展更多的临床试验,并最终成为来源于复杂天然混合物的新药,以满足目前未满足的医疗需求。 从膳食补充剂到新药如1994年美国《膳食补充剂健康与教育法案》(Dietary Supplement Health and Education Act,DSHEA) [2]所述,植物产品如有与健康相关的声明,可以以传统食品、膳食补充剂或药品的形式上市。传统食品与膳食补充剂如没有防治疾病声明,则由FDA食品安全与应用营养中心监管。根据《食品、药品和化妆品法案》,药品定义为缓解、治疗、治愈、诊断或预防疾病或其相关症状(疾病声明),或旨在对机体的结构或功能(结构—功能声明)产生影响。根据DSHEA,膳食补充剂只在声明与防治疾病有关时被认为是药品。在这种情况下,膳食补充剂由FDA药品审评与研究中心(Center for Drug Evaluation and Research,CDER)监管。植物药与非植物药申请在适用FDA法规方面通常没有差异。特别是,为了新药上市,植物药必须提供符合法规要求的安全性与有效性证据(这与《食品、药品和化妆品法案》相关部分一致),并遵循生产要求以确保产品质量。制造商根据法规膳食补充剂如果没有得到新药申请批准,则不能标明能够治疗药物。但是,许多无确实证据的植物膳食补充剂用于医疗是众所周知的,并可以在文献、新闻媒体或互联网上看到。根据DSHEA,已上市植物产品如没有防治疗疾病声明,则不要求向FDA报告临床试验。2000年1月5日(后续修订)[3],FDA法规中定义了防治疾病声明与结构—功能声明(有时称为“保健声明”)的差异,但这种差异通常较小且容易混淆。因此,膳食补充剂可以声明对“骨保健”有益,而不能说是治疗或预防骨质疏松症或骨折。然而,除制造商之外,公众能公开使用防治疾病声明。自我诊疗的消费者经常会把许多植物膳食补充剂作为药品(其有效性与安全性没有监管保证)。到目前为止,除了根据非处方药(Over the Counter,OTC)专论已上市的几个植物非处方药(例如,洋车前子与番泻叶)和洋地黄叶(其制剂以洋地黄毒甙的含量为标准),仅有Veregen一个植物产品作为新处方药得到FDA的批准。 目前美国的监管环境原则上, FDA批准所有新药时要求的产品质量标准以及有效性与安全性证据,也适用于希望在美国作为药品上市的新植物产品。在对临床数据的要求方面,植物药与非植物药没有区别,但是对于产品质量控制的要求,则根据植物药是混合物这一事实(其活性化合物可能不知道)需要进行调整。但是,监管的目的不是要在药物之外另设一类植物药分类,而是为了确保在质量和临床效果方面,与对目前已有的非植物药一样有同等程度的信心。为了鼓励与促进植物药的发展, CDER于2004年7月发布了《植物药新药指南》[1]。根据植物药的独特性以及在研制过程中遇到的实际困难(例如,复杂混合物、未知或未完全证实的活性成分和以往充分人类使用历史),CDER对初期植物药研究的要求进行了调整。为了支持进行早期的临床试验,与以前没有人用历史的合成药物相比,对植物药的非临床药理和毒理研究,以及化学、生产和控制(chemistry, manufacturing and controls,CMC)的早期要求可以明显地减少。例如,不要求申请者进一步纯化或证实植物产品的活性成分。与纯化非植物药不同,对复杂植物原料药和植物产品而言,用常规的CMC方法来控制质量可能还不充分。为确保植物药批次间的一致性,对原药材的质量控制需要附加要求。对于支持IND申请的CMC和非临床评价的要求程度取决于其以往人类使用经验、与传统剂型和用法的差异、以及申请的临床研究规模。总之,CDER鼓励申请者递交关于以往人类使用经验的所有资料用于对IND初步安全性进行评估,来决定这些经验与申请的临床研究相关性。尽管在没有非临床毒性数据支持的情况下,申请者可能对一些质量规范化且广泛使用的植物药开始进行较大规模的临床试验。但在NDA时,可能需要追加动物研究[4,5]。需要强调的是,支持植物药上市批准的安全性与有效性证据的整体标准与非植物药同样严格。值得注意的是在FDA指南[1]中,药品监管机构认为植物药与非植物药属于同一类[4,5]。它们同样需要符合严格的质量标准以及对临床数据的要求。在此方面FDA不同于欧洲(欧盟药品评价局 (The European Medicines Agency,EMEA);伦敦)和加拿大(加拿大卫生署;多伦多)监管当局。除了非植物药、食物和膳食补充剂分类之外,EMEA与加拿大卫生署已经建立另外的产品分类,比如“草药产品—基于良好数据的用法和传统经验的用法” [6]和“天然保健品” (http://www.hc-sc.gc.ca/dhp-mps/prodnatur/index-eng.php)。EMEA与加拿大卫生署可能依据以往人类使用经验(包括已上市的中药或草药用法)、专家意见或对可获得的安全性与有效性数据文献的综述,批准植物产品以药品形式上市,并表明药用范围。而根据监管当局许可进行的临床试验可能是不必须的。
大家都知道浸出工艺生产的植物油,是需要溶剂提取的,难免会有溶剂残留在其中。可是根据压榨工艺的描述应该不会有溶剂残留的存在,可是试剂情况却是有些压榨工艺的产品有溶剂残留甚至还出现含量较高的情况。并且在一些标准中对低等级的压榨工艺的产品的规定了一定的限量,却不是不得检出。不知道压榨工艺的植物油中溶剂残留的来源都有哪些呢?敬请各位版友发表一下自己的观点,谢谢!
今日环保部发布水质 石油类和动植物油类的测定 红外分光光度法(HJ 637-2012)新标准,并规定标准自2012年6月1日起实施,实施之日起原GB/16488-1996作废。本次为GB/16488的第一次修订,主要修订内容如下: ——增加了总油的定义; ——修改了无水硫酸钠和硅酸镁的处理条件; ——修改了样品体积的测量方法; ——修改了样品的萃取条件和萃取液脱水方式; ——删除了絮凝富集萃取内容; ——删除了非分散红外光度法内容。 各位测油版友针对新标准有何看法!请发表高见。
[size=16px] 叶绿素测定仪通常用于测量植物叶片中的叶绿素含量,而不是直接测量氮含量。然而,叶绿素含量与植物的氮素含量之间存在一定的关联,因为氮是叶绿素分子中的一个重要组成部分。叶绿素测定可以作为一种间接方法来估计植物的氮含量。 要测量植物的氮含量,通常可以使用以下方法之一: Kjeldahl法:这是一种传统的分析方法,可以测量有机物中的氮含量。样品首先被消化,然后氮被转化为氨,并通过滴定酸来测量氨的含量,从而计算样品中的氮含量。 Dumas法:这是一种更现代的方法,类似于Kjeldahl法,但使用燃烧而不是消化来将样品中的有机氮转化为氨。然后通过化学反应测量氨的含量,从而计算氮含量。 [url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法:这是一种通过[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]来分析样品中的氮化合物的方法。样品在高温条件下分解产生氮气,然后氮气被送入[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]进行分析。 光谱法:虽然叶绿素测定仪主要用于叶绿素含量测量,但某些光谱数据也可以用于估计氮含量。光谱数据中的某些特征可以与氮含量之间存在的关联进行校准,从而进行估算。 请注意,这些方法可能需要特定的实验室设备和技术,并且样品的处理和分析可能会有一定的复杂性。云唐建议选择合适的方法取决于你的实验目的、设备可用性以及实验室的专业知识。如果你不熟悉这些分析方法,最好是在有经验的人的指导下进行实验。[/size]
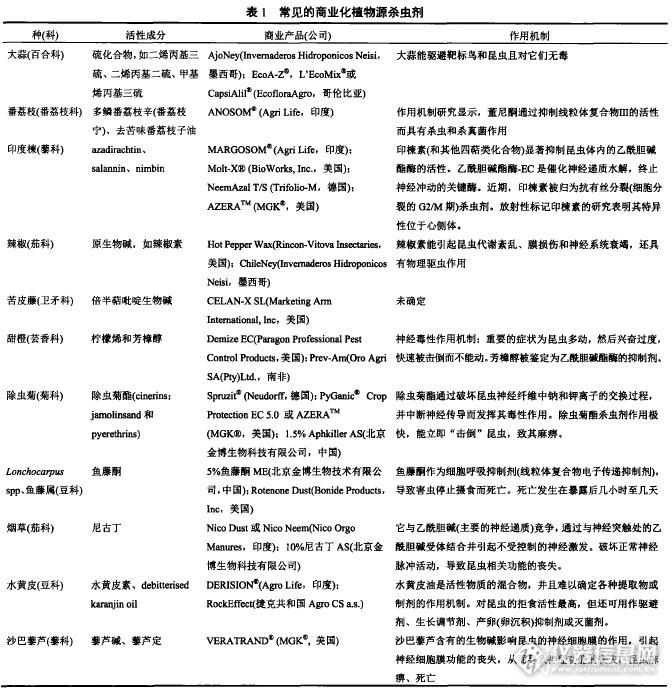
在欧洲用植物防治昆虫可追溯到3000多年前。起初,人们以一些芳香植物和其提取物或汤液防治害虫,特别是作为驱避剂对付肠虫或外寄生虫等讨厌的昆虫,也用植物来保护贮存的粮食或食物免受仓储害虫的危害。第一个商业化植物源杀虫剂出现在17世纪,当人们发现烟草叶中的尼古丁能杀死豆象虫后,开发上市。1850年左右,一种叫做鱼藤酮的植物源杀虫剂问世了,它是从鱼藤的根中提取获得。第二次世界大战之后,在欧洲廉价的合成杀虫剂(有机氯和有机磷酸盐)的出现(主要由部分化武制剂脱毒而成),使商业化植物源杀虫剂的进一步发展受到限制。http://ng1.17img.cn/bbsfiles/images/2017/03/201703261604_01_1623180_3.jpghttp://ng1.17img.cn/bbsfiles/images/2017/03/201703261604_02_1623180_3.jpg注:相关内容摘自《世界农药》
各位大虾,请问有没植物中铅含量测定的标准方法啊?
请问大家是否有《植物中营养元素检测的标准》,麻烦帮传一下!谢谢!
谁有JJF 049-2006 落锤式织物撕裂仪校准规范!不胜感激!有份校准证书看不懂! 结论: 扇形摆锤摆幅衰减量:25mm。
我是做植物激素的,希望与做植物激素相互探讨。QQ 278181880







 我要推广仪器
我要推广仪器
 下载APP
下载APP