抗癌”神药” Opdivo现严重副作用 人人适用吗?基因检测来解答
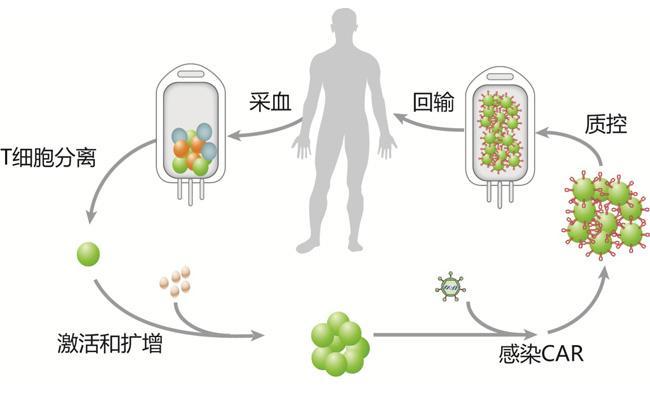
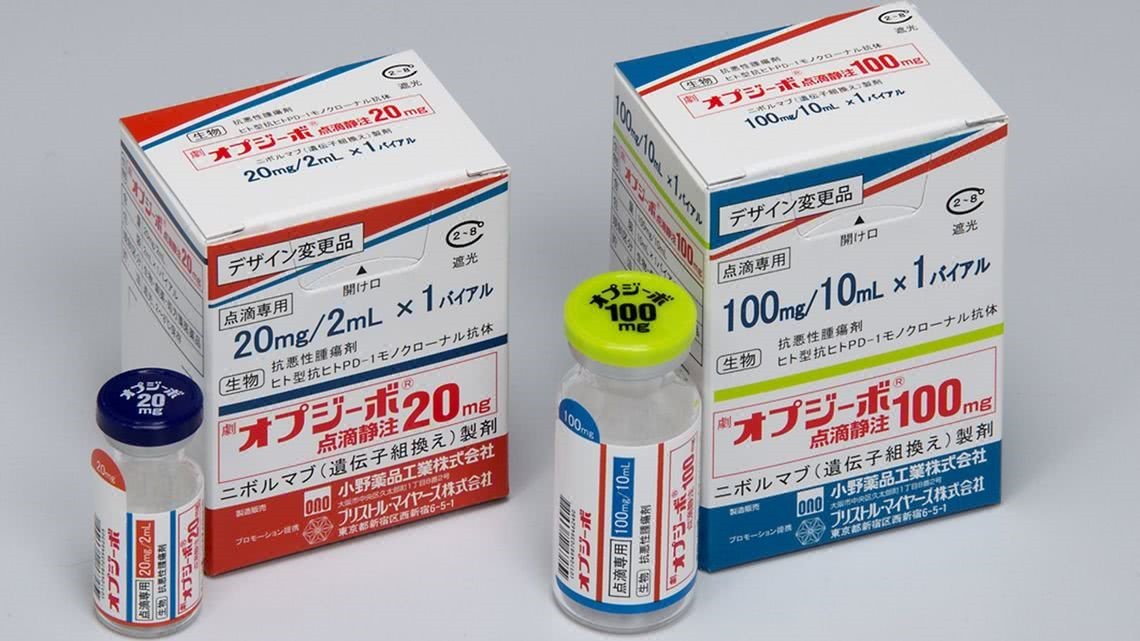
p style=" text-align: justify text-indent: 2em " span style=" text-indent: 2em " 5月9日,日本医药品医疗器械综合部门表示,目前已经有11名患者在使用opvido后发生脑部疾病,其中1人死亡。为此,日本厚生省已向小野药品工业下达指标,要求其在药品使用说明书中补充其具有的“严重副作用”。目前,小野药品工业已经在使用说明书中对Opdivo 具备的“副作用”进行补充说明,并提醒各医疗机构注意其副作用。 /span /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 409px height: 226px " src=" https://img1.17img.cn/17img/images/201905/uepic/915eb94f-d270-4c6e-8979-f97ecccb4457.jpg" title=" 001.jpg" alt=" 001.jpg" width=" 409" height=" 226" / /p p style=" text-align: center text-indent: 0em " 日本市售Opvido(东洋经济 图) span style=" text-indent: 0em " & nbsp /span /p p style=" text-align: left text-indent: 2em " span style=" color: rgb(192, 0, 0) " strong 抗癌“神药”Opdivo与诺贝尔奖 /strong /span /p p style=" text-align: justify text-indent: 2em " 对于近两年广为人知的PD-1单抗Opdivo(中文名“欧狄沃”,简称O药),该药品由美国药企百时美施贵宝和日本小野药品工业株式会社(下称小野药品)合作生产,目前已在中国上市,用于治疗非小细胞肺癌。Opdivo最初是由日本小野药品工业公司在诺贝尔奖得主京都大学本庶佑(Honjo Tasuku)教授的研究成果基础上,开发研制的一款抗癌药。 /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 405px height: 233px " src=" https://img1.17img.cn/17img/images/201905/uepic/bb589e91-ffb8-4c59-9d38-3000dd5887c0.jpg" title=" 002.jpg" alt=" 002.jpg" width=" 405" height=" 233" / /p p style=" text-align: center text-indent: 0em " 诺奖得主 本庶佑(图:东京新青年) /p p style=" text-align: justify text-indent: 2em " 据悉,目前在全球范围内共有9款PD-1/PD-L1单抗药物上市,分别为百时美施贵宝(Opdivo)、默沙东(Keytruda)、罗氏(Tecentriq)、阿斯利康(Imfinzi)、默克/辉瑞(Bavencio)、君实生物(拓益)、信达生物(达伯舒)、赛诺菲/再生元(Libtayo)、恒瑞医药(艾力妥)。 /p p style=" text-align: justify text-indent: 2em " strong span style=" color: rgb(0, 112, 192) " 对此事件,网友们发表了各自的意见和看法: /span /strong /p p style=" text-align: justify text-indent: 2em " span style=" text-decoration: underline color: rgb(0, 112, 192) " strong 网友1: /strong 这才是现代医药负责人的表现,有什么副作用一定要刺耳的清清楚楚,不可以写“尚不明确”。 /span /p p style=" text-align: justify text-indent: 2em " span style=" color: rgb(0, 112, 192) text-decoration: underline " strong 网友2: /strong 有些企业只是想分享医疗进步的红利,却不想承担医疗进步带来的风险代价。 /span /p p style=" text-align: justify text-indent: 2em " span style=" color: rgb(0, 112, 192) text-decoration: underline " strong 网友3: /strong 实验阶段干什么去了?非得上了临床给人用了才发现有这么重大的副作用! /span /p p style=" text-align: justify text-indent: 2em " span style=" color: rgb(192, 0, 0) " strong Opdivo人人适用吗?基因检测来解答 /strong /span /p p style=" text-align: justify text-indent: 2em " 生物制药引起副作用的原因极其复杂,而对于如此一款“神药”,也并非每个患者都适用。据了解,如果是实体瘤患者,需要考虑进行基因检测。国外有许多临床试验证明,一些特定的基因检测结果为阳性的患者,使用PD-1抑制剂治疗是有效的。为此,小编整理了几款在国内获得医疗器械许可的“有证”基因测序仪。 /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 276px height: 213px " src=" https://img1.17img.cn/17img/images/201905/uepic/806478c1-9546-463d-94b7-ae8fed415c5e.jpg" title=" Illumina的MiSeqDx测序仪.jpg" alt=" Illumina的MiSeqDx测序仪.jpg" width=" 276" height=" 213" / /p p style=" text-align: center text-indent: 0em " a href=" https://www.instrument.com.cn/netshow/SH103701/C241668.htm" target=" _blank" span style=" color: rgb(0, 112, 192) text-decoration: underline " Illumina的MiSeqTM Dx测序仪(点击查看仪器参数、报价信息) /span /a /p p style=" text-align: justify text-indent: 2em " MiSeq& #8482 Dx基因测序仪是 strong Illumina /strong 首款通过中国国家药品监督管理局医疗器械注册批准(2018年8月)的进口基因测序仪,意味着Illumina可以将MiSeq& #8482 Dx基因测序仪推广并销售给中国境内的医院及其他医疗机构进行体外诊断(IVD)检测。MiniSeq测序系统实现一触式的操作,直观的用户界面,快速、简单的流程并采用Illumina成熟、经典的测序技术。不但仪器具有成本优势,而且测序少量样本也具有成本优势。其直观的从样本到答案工作流程能够快速对DNA和RNA相关应用进行测序。MiniSeq有着广泛、灵活的应用,能够让研究人员在不同测序项目间灵活转换,如不同的RNA或DNA样本应用,包括信号通路分析,实体瘤和血液肿瘤的分析及生殖细胞系变异检测。优化的分析流程可适用于不断增加的新应用。 /p p style=" text-align: justify text-indent: 2em " strong 华大基因 /strong 继2014年BGISEQ-100,BGISEQ-1000,2016年BGISEQ-500、2017年BGISEQ-50、2018年MGISEQ-2000与MGISEQ-200获得CFDA认证之后,共计拥有6款已经获得医疗器械注册证的基因测序仪。 /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 237px height: 199px " src=" https://img1.17img.cn/17img/images/201905/uepic/c80c3643-2d21-48ce-82d8-54ac7422e356.jpg" title=" BGISEQ-50.jpg" alt=" BGISEQ-50.jpg" width=" 237" height=" 199" / /p p style=" text-align: center text-indent: 0em " a href=" https://www.instrument.com.cn/news/20161117/206574.shtml" target=" _blank" span style=" color: rgb(0, 112, 192) text-decoration: underline " 华大智造BGISEQ-50(点击查看相关资讯) /span /a /p p style=" text-align: justify text-indent: 2em " BGISEQ-50是一款更专注、更小巧、更精简的高通量测序平台。该系统沿用了先进的联合探针锚定聚合技术(cPAS)和DNA纳米球(DNB)核心测序技术,采用高精密部件,内置独立的样本加载试剂槽和全自动试剂针穿刺系统,在简约化的结构中自动集成样品加载、测序和分析等多项功能。BGISEQ-50体积小巧,易于安放,不仅全面支持临床领域和科研领域的基础测序应用项目,而且适用范围广泛,突破空间限制,即使在高海拔下的低气压环境中也能正常运行。 /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 333px height: 266px " src=" https://img1.17img.cn/17img/images/201905/uepic/968054dc-a439-4916-8a59-16ee1321c7f0.jpg" title=" 20190220094802_8671.jpg" alt=" 20190220094802_8671.jpg" width=" 333" height=" 266" / /p p style=" text-align: center text-indent: 2em " a href=" https://www.instrument.com.cn/netshow/C312327.htm" target=" _blank" span style=" color: rgb(0, 112, 192) text-decoration: underline " 华大智造MGISEQ-2000(点击查看参数、报价信息) /span /a /p p style=" text-align: justify text-indent: 2em " 华大智造自主研发的MGISEQ-2000是一款桌面型高通量基因测序仪,在2018年6月通过CFDA的审查,获得医疗器械注册证。MGISEQ-2000采用全新的芯片系统,在双芯片独立运行平台的基础上,支持不同规格的芯片,能够实现更全面更灵活的测序体验。平台自带初级数据分析软件,可以自动完成图像分析,并产生有质量打分的碱基序列,适用于科研、医学临床、司法、农业等领域,包括全基因组测序以及肿瘤基因热点突变检测。 /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 355px height: 285px " src=" https://img1.17img.cn/17img/images/201905/uepic/7ad59b6c-60d6-4654-a2b3-a460f947a12f.jpg" title=" 20180929174223_2617.jpg" alt=" 20180929174223_2617.jpg" width=" 355" height=" 285" / /p p style=" text-align: center text-indent: 0em " a href=" https://www.instrument.com.cn/netshow/C312558.htm" target=" _blank" style=" color: rgb(0, 112, 192) text-decoration: underline " span style=" color: rgb(0, 112, 192) " 华大智造MGISEQ-200(查看参数、报价信息) /span /a /p p style=" text-align: justify text-indent: 2em " MGISEQ-200是华大智造自主研发出的一款桌面型基因测序仪,在2018年6月通过CFDA的审查,获得医疗器械注册证。总重量仅有87KG并且占地面积小,在多样的环境中为您提供稳定而准确的测序,包括高海拔的低压环境和高风浪的海洋环境。适合靶向基因和小型基因组测序、微生物检测等,是基础科研和临床研究的好帮手。在PE100读长模式下满负荷运行不到48小时,平均一天能完成24个肿瘤样本,能在较短时间内完成完整的基因测序流程。 /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 267px height: 175px " src=" https://img1.17img.cn/17img/images/201905/uepic/ffa7b74d-3ae9-432e-9432-8017a0daea3f.jpg" title=" 华大智造BGISEQ-500.png" alt=" 华大智造BGISEQ-500.png" width=" 267" height=" 175" / /p p style=" text-align: center " a href=" https://www.instrument.com.cn/zc/134.html" target=" _blank" span style=" text-indent: 0em text-decoration: underline color: rgb(0, 112, 192) " 华大智造BGISEQ-500(点击进入基因测序仪专场) /span /a /p p style=" text-align: justify text-indent: 2em " BGISEQ-500是一款高通量基因测序平台,以联合探针锚定聚合技术(cPAS)及改进的DNA纳米球(DNB)为核心技术支撑。作为一款开放的一站式测序平台,BGISEQ-500具有精准、简易、快速、灵活、可拓等特点,全面覆盖科研、生殖健康、现代农业、病原识别、临床研究等领域的多种应用。 /p p style=" text-align: justify text-indent: 2em " 作为基因组学的核心技术,DNA测序技术是当下应用最广泛的生命科学技术之一。基因测序仪不仅用于科研,也正在成为精准医学的重要工具。基因测序技术不断推陈出新,从1986年第一台商用DNA测序仪的推出,到现在三代测序仪崭露头角, a href=" https://www.instrument.com.cn/news/20190327/482498.shtml" target=" _blank" span style=" text-decoration: underline color: rgb(0, 112, 192) " 小编在另一篇文章(详情可点击查看图集|基因测序仪全家福 /span /a )中梳理历代DNA测序平台 span style=" color: rgb(192, 0, 0) " strong 共58台 /strong /span ,为行业相关从业人员理清思路。附上部分列表: /p p style=" text-align: center" img style=" max-width:100% max-height:100% " src=" https://img1.17img.cn/17img/images/201905/uepic/b54703ac-5467-4747-9643-63856c702e85.jpg" title=" 00000.png" alt=" 00000.png" / /p p style=" text-align: center " strong style=" color: rgb(0, 112, 192) font-size: 18px text-indent: 0em " & #8230 & #8230 /strong /p p style=" text-align: center text-indent: 0em " span style=" color: rgb(0, 112, 192) " 欲免费查看完整列表,请扫码关注 span style=" color: rgb(192, 0, 0) " strong 3i生仪社 /strong /span ,回复“ span style=" color: rgb(0, 0, 0) " strong 基因列表 /strong /span ”即可。 /span /p p style=" text-align: justify text-indent: 2em " span style=" color: rgb(0, 112, 192) " /span /p p style=" text-align: center" img style=" max-width:100% max-height:100% " src=" https://img1.17img.cn/17img/images/201905/uepic/52077d41-d959-47d1-85db-f02ca150ba3e.jpg" title=" 71131a07-2b8a-4876-bfc2-f9776e680221.jpg" alt=" 71131a07-2b8a-4876-bfc2-f9776e680221.jpg" / /p p style=" text-align: justify text-indent: 2em " 如 strong span style=" color: rgb(192, 0, 0) " 网友3 /span /strong 所言,研发人员在药物研发及生产期间,就应该对其进行充分的检测,确保万无一失才能进行下一阶段应用。生物制药的分析、表征与质量控制是药物研发过程中必不可少的环节,仪器信息网特别推出了 a href=" https://www.instrument.com.cn/zt/swzybz" target=" _blank" span style=" color: rgb(0, 112, 192) text-decoration: underline " 生物制药在分析、表征与质量控制部分仪器设备与解决方案(点击查看相应专题了解更多生物制药研发必备“神器”) /span /a 。 /p p style=" text-align: center" a href=" https://www.instrument.com.cn/zt/swzybz" target=" _blank" img style=" max-width: 100% max-height: 100% width: 523px height: 181px " src=" https://img1.17img.cn/17img/images/201905/uepic/78be79eb-a426-4c96-af81-035488d33915.jpg" title=" 11111111111.png" alt=" 11111111111.png" width=" 523" height=" 181" / /a /p p style=" text-align: justify text-indent: 2em " 期待科学家们在生物制药副作用方面的研究取得更多进展,让精准医疗真正实现精准用药、无副作用!仪器信息网小编也会持续关注此次事件后续报道。 /p p style=" text-align: center " span style=" text-decoration: underline " & nbsp & nbsp & nbsp & nbsp & nbsp & nbsp & nbsp & nbsp & nbsp & nbsp & nbsp & nbsp & nbsp & nbsp & nbsp & nbsp & nbsp & nbsp & nbsp & nbsp & nbsp /span br/ /p p style=" text-align: center " span style=" text-decoration: none " 号外号外!关注 strong span style=" text-decoration: none color: rgb(0, 112, 192) " 仪器信息网 /span /strong 生命科学公众号“ span style=" text-decoration: none color: rgb(0, 112, 192) " strong 3i生仪社 /strong /span ” /span /p p style=" text-align: center " 解锁更多生命科学仪器资讯 /p p style=" text-align: center " img style=" max-width:100% max-height:100% " src=" https://img1.17img.cn/17img/images/201905/uepic/22109573-c2e3-491d-8710-bd5ee138d63b.jpg" title=" 71131a07-2b8a-4876-bfc2-f9776e680221.jpg" alt=" 71131a07-2b8a-4876-bfc2-f9776e680221.jpg" / /p p style=" text-align: center " img style=" max-width:100% max-height:100% " src=" https://img1.17img.cn/17img/images/201905/uepic/f8feb7e5-9636-4fc2-8cac-23538099e472.jpg" title=" 企业微信截图_20190520102956.png" alt=" 企业微信截图_20190520102956.png" / /p
























 我要推广仪器
我要推广仪器
 下载APP
下载APP