泵压力不稳定,而且经常抽不上流动相。会是什么原因呢?没有地方漏液
大家都知道,流动相抽滤装置主要用于HPLC流动相的过滤,并具有一定的脱气作用,以保证流动相的洁净,防止HPLC液路的堵塞。那么,在工作中,你们用来抽滤过其它的物质吗?

[align=center][b]高效液相色谱仪故障分析-泵无法正常抽吸流动相[/b][/align]高效液相色谱系统硬件一般很稳定,一套安捷伦系统甚至在合理使用的情况下,使用10年依旧可以保证保留时间的稳定。而新手在使用高效液相色谱系统时,对部件不了解导致各个部件容易出现故障。其中液相色谱系统中,色谱泵无法正常抽吸流动相是困扰各位老师的常见故障之一。本文为大家介绍解决办法及措施,遇到类似问题再也不用麻烦工程师啦![img=,690,920]https://ng1.17img.cn/bbsfiles/images/2019/07/201907241648112426_1300_3922638_3.jpg!w690x920.jpg[/img][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/07/201907241648319487_9751_3922638_3.jpg!w690x517.jpg[/img]泵无法正常抽吸流动相,典型的表现为流动相试剂瓶中有小段空气,设置泵流速后,空气在管路中徘徊,无法抽吸流动相,这时大家应卸下管路与泵头连接的旋钮,将流动相管路中气泡排出。检查试剂瓶中没有出现负压现象的前提下,观察试剂瓶中的流动相是不是乙腈。[img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/07/201907241648485262_7281_3922638_3.jpg!w690x517.jpg[/img] 乙腈经常放置于棕色试剂瓶,其原因是在光照下乙腈分子直接容易产生黏连,而泵无法正常抽吸流动相,正是乙腈黏连将色谱泵堵塞,导致色谱泵无法正常抽吸流动相。此时不需要将泵头拆除,只需要将泵头前的单向阀拆下,超声10分钟即可。下图是色谱泵各部件的示意图。[img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/07/201907241649140208_3906_3922638_3.jpg!w690x517.jpg[/img][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/07/201907241649223426_5553_3922638_3.jpg!w690x517.jpg[/img]
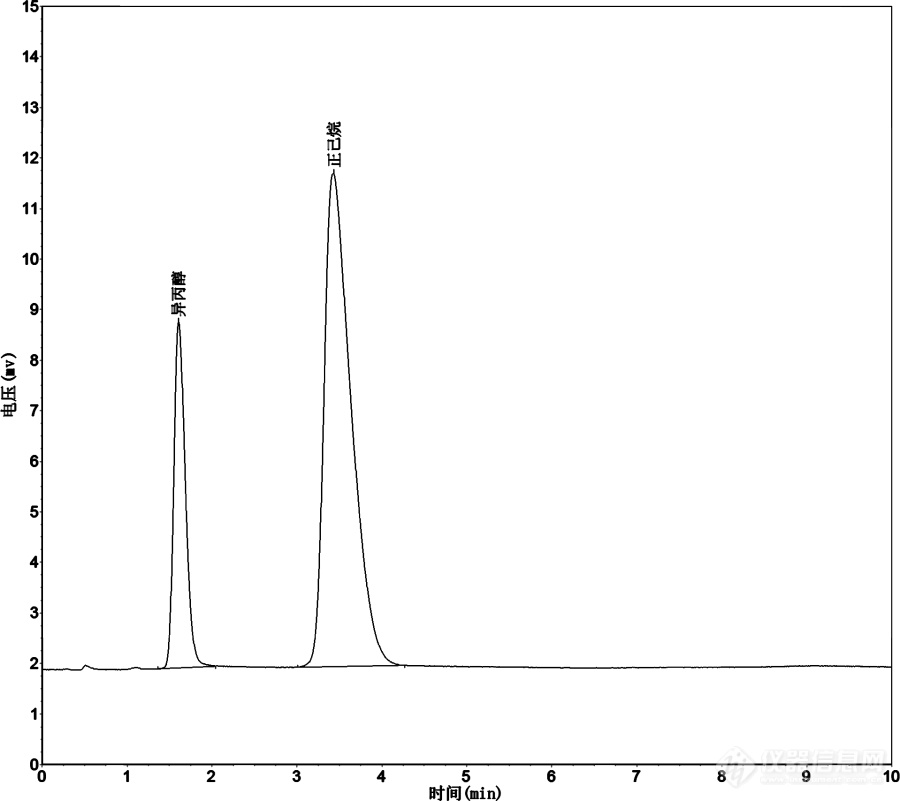
[b]1、问题的提出[/b]液相色谱的流动相不应该含有固体微粒和溶解气体,因此过滤和脱气应该是液相色谱实验员最常规的日常工作。即使现在普遍购买色谱级的溶剂,并且很多仪器都配了在线脱气机,通常还是推荐流动相要先过滤和脱气。.流动相的过滤通常采用的是负压抽滤以提高效率,而脱气最简便常用的方式是超声。[b]这里就引出了一个长期争论不休的问题:是先按比例混合流动相然后抽滤和超声,还是先分别抽滤和超声之后再混合?[/b]我的习惯是先混合,然后抽滤和超声,因为两种或多种溶剂混合后,由于极性改变,溶解的固体会再次沉淀出来、溶解的气体也会析出产生新的气泡。在配置水-甲醇混合流动相的时候就是非常明显的,对于分别抽滤和超声处理好的水和甲醇,一混合马上就会产生大量气泡,如果不再次超声就很容易导致气泡吸到泵里去。但是也有人认为应该分别抽滤和超声,然后按比例混合,其理由是:不同溶剂的挥发速度有差异,抽滤和超声导致溶剂挥发损失的同时,流动相的组成就会偏离初始配置的组成。这种观点也是很有道理的,比如水挥发很慢,而低沸点的乙腈挥发明显快得多,而弱极性且沸点低的溶剂,比如正己烷、二氯甲烷等,挥发就更快了。一些权威的专著也是支持这一观点的(例如:色谱技术丛书第二版,于世林编著《高效液相色谱方法及应用》,第13页)。那么,到底应该采取哪种做法呢?按比例混合后再进行抽滤和超声,是不是真的会改变流动相的组成?本着实践出真知的原则,我们来实际测一下吧。.[b]2、实验测定实验内容:[/b]选取常见流动相中挥发能量比较强的正己烷进行考察,添加的第二组分为异丙醇。异丙醇沸点较高且极性较强,挥发性比正己烷弱很多。因此这一体系将会比较容易体现出差异。按正己烷/异丙醇=8/2的比例配置流动相500mL,用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测定其相对含量;然后抽滤(-0.7atm,约5min),再次进行[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定;然后放置在500mL玻璃试剂瓶中(不加盖)超声10min,进行第三次[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定。实验室气温为28℃。.[b]测定方法:[/b]Hayesep-D填充柱,2mm*1.5m,氢气为载气,流速15mL/min,柱温220℃。TCD检测器,桥电流80mA,温度150℃。进样口150℃,进样0.2uL。典型色谱图如下:[img=,690,614]https://ng1.17img.cn/bbsfiles/images/2020/07/202007242145015251_9814_2204387_3.png!w690x614.jpg[/img]采用面积归一化法进行定量。由于没有考虑校正因子的差异,面积百分比与真实的流动相组成有一定误差,但是这属于系统误差,用于比较前后变化时不会存在问题。.[b]3、结果与讨论[/b]话不多说,直接上实验结果:[img=,690,412]https://ng1.17img.cn/bbsfiles/images/2020/07/202007242151219077_7807_2204387_3.png!w690x412.jpg[/img]原始流动相中异丙醇的测定值为22.79%,经过抽滤和超声之后依次变为23.10%和23.07%,可见有一定变化。通过t检验法检验差异的显著性。抽滤前后计算得:t=12.9,远大于置信度99%时的临界值,说明抽滤导致的组成变化是显著的。正己烷损失更多,导致异丙醇相对比例提高。超声前后计算得:t=1.4,,原小于临界值,说明超声导致的变化不显著。.但应该注意到,抽滤前后,流动相组成的变化仅为0.31%,而平时用量筒手工配置流动相时,准确度做到正负1%以内就已经不错了,即使是通过比例阀在线混合,准确度也很难达到0.1%量级。因此可以认为,抽滤导致的流动相组成变化的问题客观存在,但对实际操作的影响却不大,属于可以忽略不计的误差。而且这里选取的是极易挥发的正己烷体系作为示例,也只表现出了0.3%左右的变化,若使用都挥发较慢的甲醇-水体系,可以预见,产生的差异可能会更小。通常采用隔膜泵进行抽滤时,真空度普遍不到-0.85atm,抽滤时间也不会太长,一般只有数分钟,这种情况下完全可以忽略抽滤导致的流动相组成变化。因此我认为先混合,然后进行抽滤是可靠的,不用担心组成变化问题。当然,如果使用沸点极低、挥发极快的二氯甲烷这类溶剂,或者遇到抽滤时间很长这种极端情况,这一问题还是需要考虑的。.[b]4、结论(1)抽滤操作过程中,易挥发组分损失较多、难挥发组分损失较少,会导致流动相组成的显著变化。超声操作对流动相组成的影响不明显。(2)抽滤操作导致的流动相组成变化一般在千分之几数量级,小于通常配置流动相的误差,因此在常规操作中都可以忽略不计。除了少数极端情况外,先混合再抽滤的操作是可靠的。[/b]

[align=center][b]高效液相色谱仪故障分析-泵无法正常抽吸流动相[/b][/align]高效液相色谱系统硬件一般很稳定,一套安捷伦系统甚至在合理使用的情况下,使用10年依旧可以保证保留时间的稳定。而新手在使用高效液相色谱系统时,对部件不了解导致各个部件容易出现故障。其中液相色谱系统中,色谱泵无法正常抽吸流动相是困扰各位老师的常见故障之一。本文为大家介绍解决办法及措施,遇到类似问题再也不用麻烦工程师啦![img=,690,920]https://ng1.17img.cn/bbsfiles/images/2019/07/201907251938215540_9623_3922638_3.jpg!w690x920.jpg[/img]泵无法正常抽吸流动相,典型的表现为流动相试剂瓶中有小段空气,设置泵流速后,空气在管路中徘徊,无法抽吸流动相,这时大家应卸下管路与泵头连接的旋钮,将流动相管路中气泡排出。检查试剂瓶中没有出现负压现象的前提下,观察试剂瓶中的流动相是不是乙腈。[img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/07/201907251938435710_7747_3922638_3.jpg!w690x517.jpg[/img] 乙腈经常放置于棕色试剂瓶,其原因是在光照下乙腈分子直接容易产生黏连,而泵无法正常抽吸流动相,正是乙腈黏连将色谱泵堵塞,导致色谱泵无法正常抽吸流动相。此时不需要将泵头拆除,只需要将泵头前的单向阀拆下,超声10分钟即可。[img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/07/201907251938547040_6003_3922638_3.jpg!w690x517.jpg[/img]下图是色谱泵各部件的示意图。Agilent1260色谱系统的色谱泵主要由密封金垫、冲洗阀、出口单向阀、入口单向阀等组成,还包含了混合器、阻尼器、压力传感器和缓冲毛细管等。系统耐压高达600 bar,使用不锈钢加固的单向阀、冲洗阀和主动阀,标准配置延迟体积为600-800微升。[img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/07/201907251939086455_9815_3922638_3.jpg!w690x517.jpg[/img][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/07/201907251939132295_6922_3922638_3.jpg!w690x517.jpg[/img]
新手上路,一台比较老的液相色谱仪,型号LC-100,手动进样,流动相水:甲醇=90:10,水的A泵正常,甲醇的B泵,工作显示正常,就是抽不上,空了一段。是不是滤头堵塞了,怎么处理?1、空的管子怎么填满流动相?2. 基线锯齿状怎么回事?请各位高手不吝赐教。
一直以来,我抽滤头经常破损,我的抽滤头与抽滤瓶磨砂口之间没涂硅油之类润滑剂,抽滤完流动向后,抽滤头与抽滤瓶常连接很牢,难以拆卸,经常借助外力,于是常常损坏,看看大家有什么好方法,使其易于拆卸而不损坏?http://image1.nowec.com/2011/2/24/bianchun520/1/5-1032383-16408213.summ.jpg
有谁知道熔体流动速率测试仪检定规程和用什么仪器来检定。
一个吸滤头坏掉了,请问大家可以拔掉直接用管子抽吸流动相吗?会不会有什么影响?
为了方便大家,做一个链接:有人担心泵把流动相抽干后会排干色谱柱,会使系统中充满气泡。这有可能吗?有人不小心拆卸色谱柱后忘了拧上封口螺钉,会造成什么后果?如果色谱柱中进入了气泡或部分干枯该如何处理?大家多次讨论过这个问题。这个幻灯片讲解的是一个老师上课教材中的答案。[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=21663]色谱柱中的流动相会排干吗?[/url]ppt文档,用rar解压后使用。
我们在抽流动相的过程中,泵的压力达到0.8MPA,已经是很大的压力。因为用的是水循环抽滤泵,用小的真空泵根本无法抽滤,因为太慢了,几乎是一滴一滴的往小滴的,咨询了抽滤膜的厂家说这样好,说明抽滤膜的密封性好,把脏东西都抽下来了,不知道是不是忽悠我们的,其它东西一切正常,现在就是没有换别的厂家的膜试过,这样的话太耽误工作了。抽滤一瓶要一个小时,急用的时候根本没有办法
流动相抽空了,怎么办?应该采取哪些措施?
流动相抽空了,压力为0,应如何处理?流动相抽空会导致什么后果?以后使用时,检测一种维生素,出峰时间越来越早,能提前10分钟左右,这个情况是流动相被抽空过导致的吗?
[color=#444444]昨晚忘记设置自动关机,流动相抽干了,今天脱气之后,用甲醇大流速的冲了柱子(苯基柱),看基线稳定了就换流动相(带缓冲盐)进样了。结果每个色谱峰都分裂了,换别的柱子就没事了。问问大家,是不是原来的那个柱塌陷了,还能补救吗?怎么补救呢?[/color]
[size=4]请问[u][b]熔体流动速率仪校准/检定机构[/b][/u]哪里有?最好是四川的,最好付上电话、地址。谢谢各位了[b]!(急)!!![/b][/size]
有时向流动相水中加一定比例的盐,请问盐的作用是什么,盐的种类都有什么,浓度常见是多少
我想把山梨醇和木糖醇分离开,可用35%的乙腈却无法稳定基线。我很少用乙腈为流动相,有高手可以帮忙吗?
今天早上发现流动相抽空,样品还没有进完,重新启动仪器和工作站,purge后发现正常流速下压力很小(阀门已关),检查管道发现有很多的气泡,继续purge,之后压力仍然没有上来,因为是四元泵就把混合后的那路管道给拆下来用针头进行抽滤后有液体流出,想着气泡应该没了,purge后放在正常流速上发现基线很是不稳,波动太大,想想偶尔一次抽空应该不会这样吧,就电话工程师,说把流通池拆下来,看看基线是否平稳,意思是有可能是灯的问题,按照工程师的意思做了之后发现基线依然不稳,当时就想灯才用不久呀,想着想着突然发现我的灯没开,等开灯之后一切正常,折腾了半天居然是灯没开,唉。。。。。。
溶剂过滤头没事A泵抽不上流动相,开排气,大流速,用针头在排废液的地方抽,也抽不出来.......其余三个泵都挺正常的,请问大概哪里坏了啊?在线等,有点急T.T
高效液相抽干了流动相怎么办?
遇到非常困惑的问题,前段时间真空泵抽滤的管子可能密封性不好,砂芯抽滤压力一直是-0.04作用,今天换了根新管子密封性比较好,压力达到了-0.09,抽滤的速度也明显上去了,但是匪夷所思的是进样的保留时间居然延后了六分钟,流动相的配比绝对没有问题,我连续重新配置了三次,都是同样的情况!简直无语了!流动相为乙腈42:甲醇27:水31:36%乙酸 0.01,不至于是因为抽滤时间的变化导致乙酸挥发量的变化引起PH变化吧?这个保留时间变化也太大了!!如何解决啊?
请问混合流动相使用前怎样处理?超声(多长时间)?抽真空?等?例如乙腈水。
过滤流动相的抽滤瓶被吸住了,原因是甲醇流到接头那部分去了,试过加热,冷却,超声,都没有能 分开, 请问大家有什么妙招吗?谢谢
[color=#444444][url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]由于对流动相要求较高,配置水相如20mM 醋酸铵溶液,采用HPLC级醋酸铵及超纯水配置,一般还要0.22um滤膜抽滤。[/color][color=#444444]仔细想想,这种HPLC级的无机盐(sigma)纯度99%及超纯水配置的水相,[/color][color=#444444]抽滤是否有必要呢?[/color]
[font=&]清洗柱子问题,先用20%乙腈冲洗了一天,然后逐渐过渡到100%乙腈,但是升高乙腈比例,压力先下降然后再缓慢升高,都是纯的流动相,这是怎么回事呢,(由于抽滤瓶放置过久,现在使用 抽滤瓶过滤的流动相背景很高,没有找到管用的清洗抽滤瓶的方法,所以流动相是没有经过过滤的,水是超纯水,乙腈是色谱级的),希望了解这方面知识的朋友可以帮忙解答一下,谢谢[/font]
如果没有文献可以查的话,怎么定流动相!由于工作需要,要测定一种新的物质。没有文献可以查,想定测定的流动相。我就是参考了,其他相近的物质,然后用TLC展开,如果分离好的话,就上机做,看看。就不知道,行不行。有点不敢,或许没有这样做过!有点怕!不知道,要定流动相是不是这样的流程啊!呵呵!!!!!!!!!!!!!!1
最近做液相,遇到的问题比较多,请各位大虾指点迷津。我想请问一下,标准品溶解后,用30:70的流动相定容。后来摸索条件发现,18:82的比例最合适。那先前用那个比例流动相定容配的标准储备液还能用不?定容用的流动相比例与实际进样用的流动相比例不同,是否会对实验结果造成较大的误差?谢谢!
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=97803]在线滴定技术_连续流动滴定的应用[/url][img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=97802]在线滴定技术_连续流动滴定的应用[/url]
我在前面问过一个问题是:在使用制备色谱时,紫外检测仪已稳定,为何流动相通过紫外检测仪时,紫外检测仪读数却不停的变动(流动相没有气泡)?专家回答有两种可能:电路不稳或流动相不纯。我的流动相是分析纯的甲醇100%,检测仪在不通流动相时是稳定的,因此两种可能排除。希望各位再帮帮我的忙,还会有什么情况?
使用四氢呋喃做流动相测分子量,在购买时有带有稳定剂BHT和不加稳定剂的,哪一种比较好?如果选用带稳定剂的,这个稳定剂会影响到塑料分子量的测试吗?







 我要推广仪器
我要推广仪器
 下载APP
下载APP