waters液相色谱,不知道怎么用紫外检测器来扫描目的物的吸收波长,帮帮我吧各位高手。[em09509]
我用的是TSB肉汤培养基作为对照,400-800nm扫描后没有吸收峰,并且样品的吸收值一直在0.2-0.8范围之中,做了四五个稀释度都是没有吸收峰。 细菌的OD值测定用的都是OD600嘛?还是不同的细菌吸收峰不一样?我扫描不出吸收峰的问题出在哪里?
各位高手们帮帮忙,今天在使用普析TU-1901进行光谱扫描时,没有找到最大吸收峰,按照文献资料其最大吸收峰应该在420nm,可是我没有扫描到那样的结果。还有就是标准曲线Abs=f(c)和f(c)=Abs的区别是什么
求助各位,我检某制剂时,标准中鉴别项规定其紫外扫描,在315,279等几处有最大吸收波长,我扫时315处有峰,但是检出峰值却不显示315的峰,请各位高手帮帮忙,为什么会有这种情况呢?怎么解决呢??急!!!!!!!帮帮忙吧!谢谢!!
原子吸收分光光度计灵敏性调节T90不大于3秒在哪调节?
史上最全面的仪器参数解读之第一集:原子吸收参数全解析主活动贴及见:http://bbs.instrument.com.cn/shtml/20101029/2892886/奖励措施:1.参与讨论的具有积分奖励 2.每个自然月评选出一位:月度参数解读之星,奖励50个积分3.所有参数解读完毕后,评选出一位:终极参数解读之星,奖励100个积分;第二名奖励80个积分,第三名奖励50个积分奖励规则:1.参与次数越多奖励越多2.参与次数最多的版友评选为参数解读之星,参与次数相同时按照得分多少评选=============分================割===============线=========================== 原子吸收参数分享1. 光学系统检测器波长范围:189~900nm;185.0~900.0nm光栅面积:≥64x72mm;光栅刻线:≥1800线/mm;狭缝:狭缝的宽度与高度均可自动选择;双闪耀波长:236nm,597nm, 全波长范围内光通量均衡;谱带宽:0.2,0.7,1.3,2.0nm(4段自动切换)2. 石墨炉系统温度:室温~2600℃,可实现低温原子化;斜坡时间:0~99秒,最小增量1秒;保持时间:0~99秒,最小增量1秒。石墨炉自动进样器指标:线性相关系数:≥0.9995。3. 操作软件技术指标数据扩展:仪器吸收值、浓度或发射强度等读数可在0.01至100倍的范围内扩展。积分时间:可按0.1秒的增量在0.1至60秒之间任选;读数方式:包括时间平均积分、峰面积和峰高测量功能;校正曲线:多达15个标准点的各种校正曲线法供选择---------------分-----------------割----------------线-------------------------由您来解析:第一部分:1.波长范围的大小影响样品的检测吗?189nm、185nm的不同会给样品检测带来区别吗?2.光栅面积、刻线、狭缝到底代表了什么意思呢?选仪器时我需要注意这些吗?3.双闪耀波长是指什么?4.谱带宽又代表了什么意思呢?0.2,0.7,1.3,2.0nm(4段自动切换)有什么优势吗?第二部分:1.选购仪器时,对于石墨炉中的最高温度有要求吗?2.何为斜坡时间?何为保持时间?题目中的0~99秒,最小增量1秒,是不是一般仪器都能达到啊?第三部分:积分时间可按0.1秒的增量在0.1至60秒之间任选,代表的是什么意思呢?你还有哪些参数有疑问?我提出的问题中有什么纰漏或错误?欢迎您回帖讨论!!!http://simg.instrument.com.cn/bbs/images/brow/em09505.gif
石墨炉原子吸收光谱仪与火焰原子吸收光谱仪都属于原子吸收光谱仪,由光源、原子化系统、分光系统和检测系统组成。 主要区别在: 1、原子化器不同 火焰原子化器:由喷雾器、预混合室、燃烧器三部分组成。特点:操作简便、重现性好。 石墨炉原子器:是一类将试样放置在石墨管壁、石墨平台、碳棒盛样小孔或石墨坩埚内用电加热至高温实现原子化的系统。其中管式石墨炉是最常用的原子化器。 原子化程序分为干燥、灰化、原子化、高温净化 原子化效率高:在可调的高温下试样利用率达100% 灵敏度高:其检测限达10-6~10-14 试样用量少:适合难熔元素的测定 2、操作条件的选择 火焰燃烧器操作条件的选择(试液提升量、火焰类型、燃烧器的高度) 石墨炉最佳操作条件的选择(惰性气体最佳原子化温度) 3、精确度 火焰原子吸收光谱法可测到10-9g/ml数量级 石墨炉原子吸收法可测到10-13g/ml数量级 4、火焰原子吸收除了其优异的性能之外更添加了在线稀释装置和可切换的真实单,双光路光学系统。 石墨炉原子吸收采用横向加热石墨管, 加热速度可高达3800K/秒, 可设置多达30个加热步骤以适合各种应用。
参照《水质 氨氮的测定 纳氏试剂分光光度法》测定水中的氨氮,显色正常,但进行光谱扫描没有发现吸收峰,为什么会这样呢?纳氏试剂的配制是碘化汞-碘化钾-氢氧化钠溶液
“光谱仪”和“分光光度计”是同一类仪器,但是“光谱仪”的名称之前是不需要冠之以“分光”的,因为要想得到光谱,就必须分光。光度计可以是积分光度计(光强计),不需要分光;一旦分光,它就是“光谱仪”。另外,“光谱仪”和“分光光度计”的结构区别是:“光谱仪”分光不需要扫描(如CCD光谱仪),工作速度快;“分光光度计”分光需要扫描,工作速度慢。分光光度法是通过测定被测物质在特定波长处或一定波长范围内光的吸收度,对该物质进行定性和定量分析。 常用的波长范围为:(1)200~400nm的紫外光区,(2)400~760nm的可见光区,(3)2.5~25μm(按波数计为4000cm~400cm)的红外光区。所用仪器为紫外分光光度计、可见光分光光度计(或比色计)、红外分光光度计或[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光光度计。[color=#00008B]注:以上资料均摘自网络,Jese.[/color]
请教各位大侠,本人需要建立一个紫外的总含量测定方法,现扫描的全波长图如下。 吸收值大一点的为药材的吸收图,小一点的为标准品的图。 由于加入的显色剂非常大量,造成200-400段的吸收值波动非常大,但是均在427nm处有一个吸收峰。 唯一不同的是,药材在479nm处也有一个吸收峰,而标准品没有。现在想选择427nm作为一个检测波长。但是有同学说,此峰峰形并不是太好,也不对称,是一个尖的峰。所以求助各位大侠,帮忙看一下,这种情况,能不能选择这个峰作为检测波长,如果不能,有没有什么方法调整峰形。注:药材和标准品在400-700段是没有吸收的,在紫外下是有吸收。

用原子吸收测铁元素时,波长仍为238.40nm,在仪器经过维护后,扫描出来的波形却完全不一样.原来应该是三个完整的波形,后来却只扫描出两个且不太对称的波形,会是什么原因引起的呢?虽然后来扫描出来波形不好,但仍能进行水样测量~~标准曲线线性和插标的结果均达到要求,但是想请教解决扫描波长时出现的问题,谢谢~~http://ng1.17img.cn/bbsfiles/images/2013/05/201305262041_441666_2539327_3.jpghttp://ng1.17img.cn/bbsfiles/images/2013/05/201305262041_441667_2539327_3.jpg
[color=#444444]HPLC检测样品显示在200~300nm处有两个最大吸收峰,但是把流分(纯度不高)收集后UV全波段扫描,结果显示基本没有吸收,哪位大神能不能解释下为什么?我是不是应该把收集的流分浓缩一下,加大浓度再全波段扫描?[/color]
请教各位,[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]波长扫描现在寻不到峰,怎么处理?谢谢图1:铅的波长扫描图“自动能量”后,“负高压”将增大,而我之前的“负高压”不到180V能量就能达到100%。图2:隔的波长扫描图三次自动扫描后,负高压到了250V,之前的负高压在180V左右。图3:铅的波长扫描图负高压200V了,经过自动能量,负高压还是高了点。图4:铅的波长扫描图现在的问题是,今天上午扫描波长有行面4幅图的样子,下午铅、隔波长扫描均寻不到峰,自动增加负高压到300V以上,仍没有峰,灯位、对光均正常。图5:系统复位提示“复位有误”,或者一直处于“请等待”。如何处理,请各位指点。
我在用aglient 1100做分析时,看见vwd的检测器可以对一种物质进行最大吸收波长扫描的说明,但我不知道如何进行? 请问各位高手能不能把整个过程详细的说明一下,谢谢了!!!!!!
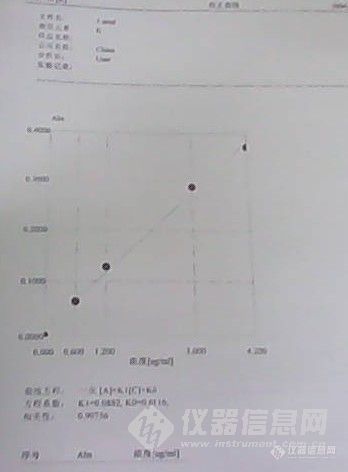
原子吸收分光光度计测定K方法改进一、问题描述:在标准NY/T889-2004中规定,土壤中速效钾和缓效钾用火焰光度计测定,随着科技的发展,由于火焰光度计测试的局限性,它逐步退出了分析测试的舞台,慢慢由原子吸收分光光度计代替。原子吸收分光光度计在测定土壤中速效钾和缓效钾时,表现出分析速度快、结果更加准确、分析方法简单等特点的同时,也有着本身的缺陷。比如,原子吸收分光光度计在测定K时,由于在空气乙炔焰中部分电离,使得溶液吸光度很大,这样无形中使得K的测定线性范围很窄、线性差、分析方法繁琐等不利于测定工作的进行。二、仪器工作条件:波长(nm)光谱带宽(nm)灯电流(mA)滤波系数积分时间(秒)766.52.0 2.0 1.0 3燃烧器高度(nm)火焰类型Air(Mpa,mL /Min)C2H2(Mpa,mL /Min)N2O(Mpa,mL /Min)5Air-C2H20.220.05,1800 三、改进:众所周知,原子吸收分光光度计在测定时是测定某元素发光强度的吸收程度。把燃烧头偏转一定角度,使得仪器只是测定部分吸收,这样可以使得原子吸收在测定时测得较小的吸光度,得到更好地标准曲线,更利于K的测定。如何偏转,如图所示:http://ng1.17img.cn/bbsfiles/images/2012/08/201208151105_383968_2352694_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/08/201208151106_383969_2352694_3.jpg改进结果:偏转前后吸光度及标准曲线偏转前浓度(ug/ml)Abs 偏转后浓度(ug/ml)Abs0.000 0.0020.000 [align
原子吸收光谱分析法在无机元素微量和痕量分析中占有极为重要的地位,也是光谱分析中中最主要的分析仪器,其应用在地矿、冶金、环境检测、医疗、商检等行业及大专院校和科研院所里得到极为广泛的应用。目前各大生产原子吸收的厂家在技术上各有优势,国内火焰法分析精度也可以与国外仪器抗衡,但总体来说国外厂商在仪器自动化、背景校正技术、石墨炉原子化、火焰原子原子化改进(原子捕集)、连续光源及仪器革新技术方面的发展比国内的势头要好,当然了不同的层次有不同的用户,不同的用户有不同的选择,只要物尽其能,人尽其力,我觉得就不错了,这是我的观点。 对于原子吸收的采购,个人认为首先应该明白下面几个问题:你是用原子吸收做普通分析还是做研究(考虑机子的档次)?做什么行业的样品(考虑测定的基体)?要分析样品里的什么元素(考虑AAS测定的方式)?样品里的被测定元素含量范围是多少(考虑测定的准确性和选择)?领导给你准备了多少money(考虑机子的范围)? 在知道了上面的内容后就可以向厂家要仪器样本(仪器样本的内容很有讲究的,大家一定要注意其中的名堂哦,有的厂家故意模糊概念、夸大其辞、隐含弊端,因此,对于不了解的方面必须要通过各方面渠道来获取可靠信息或通过合同来约定法律责任)了。详细了解各厂家的仪器样本后,可以通过其他途径(仪器用户、论坛等)来了解你感兴趣的型号,确定大体的机型范围,再拿自己的标准样品走访仪器厂家的分析室(如果条件许可,可以随同他们的检验人员观测一下仪器的测定过程,有些仪器样本描述里不太明白的东西可以向他们咨询,亲身体会哦,很重要的),在经过亲身经历后,就可以根据仪器厂家分析结果的准确性和自己的喜好进行决定性选择了。下面着重谈谈在普通分析用户采购原子吸收光谱仪时本人认为需要注意的几个方面: 注:下面所描述的仅是单方面的性能,而一台完善的原子吸收需要来看其整体性能的设计的是否平衡,应用人员的知识层次,因此,在采购原子吸收时大家可以带着这些问题去做实地的考察和样品测试过程,选择适合自己的就是最完美的。由于水平有限,错误纰漏之处难免,希望同行的朋友不吝指教。1.光路系统: 光路系统应主要了解系统的光源和光源分布、单色器结构、色散元件的性能、波长扫描及性能、光谱带宽、检测器性能。1.1光源和光源分布: 原子吸收光源主要是空心阴极灯、无极放电灯、连续光源,制造空心阴极灯的技术比较成熟,没有什么太大问题,而无极放电灯目前只有砷、铋、镉、铯、铷、锗、汞、磷、铅、钙、锑、碲、硒、钛、锌几种元素的,相对于各元素对应的空心阴极灯具有背景小、发射强度大、光源干扰少的优点,但其成本也高,至于连续光源是最新发展的技术,要配合其他部件才能发挥其强大的功能。总体来说做为光源要求高强度,高稳定性,干扰少。采购需要注意的是测定砷、汞、铋、锑等用空心阴极灯测定时灵敏度低的元素最好选用无极放电灯。光源分布简单的说就是空心阴极灯架(连续光源不考虑这个问题)的结构,现在一般的原子吸收光谱仪都具备了至少两个灯架,有的多达8个,灯多,一次予燃,可以减少测定过程中等待空心阴极灯预热的时间,其实就这么点优点,不过VARIAN AA280FS采用了快速序列技术,据说可以达到单道扫描ICP的分析速度。在设计中有的采用固定灯架,有的采用可移动的灯架。需要说明的是个人觉得采用灯架固定的比较好,因为低熔点元素的灯在预热的情况下来回转动可能损坏空心阴极灯,还要注意选用对灯的调节要比较方便好使的,当然了如果能有软件自动调节最佳位置和设置参数的更好,这个主要是考虑资金和使用者自己的情况来确定,另外对分析需要无极放电灯用户,要考虑有无极放电灯的灯架。BCC:G8 1.2单色器结构: 主要有Ebert型(如热电S系列、GBC等),C-T型(应该是Ebert型的一种改进)(如华洋、普析、瑞利、上海精密、岛津、VARIAN、北京瀚时CAAM-2001、 JENA VAVIO 6、ZEEnit60/700、日立的等),Littrow型(如PE6/7/800的等),Echelle型(以大色散为著称,如JENA ContrAA、PE的SIMAA6000、热电M系列等)。其中C-T型即水平对称设计的,比较多,由于准直镜的象差被成像物镜抵消,因此可以消除象差影响;[color=#0f
首先明确一下什么是末端吸收:指的是流动相在紫外短波段的吸收。描述一种溶剂的末端吸收常用其截止波长描述。溶剂的紫外截止波长指当小于截止波长的辐射通过溶剂时,溶剂对此辐射产生强烈吸收,此时溶剂被看作是光学不透明的,它严重干扰组分的吸收测量。其测量是将溶剂装入1cm的比色皿,以空气为参比,逐渐降低入射波长,溶剂的吸光度A=1时的波长称为溶剂的截止波长,也称极限波长。乙腈的截止波长是190nm,而甲醇是210nm。
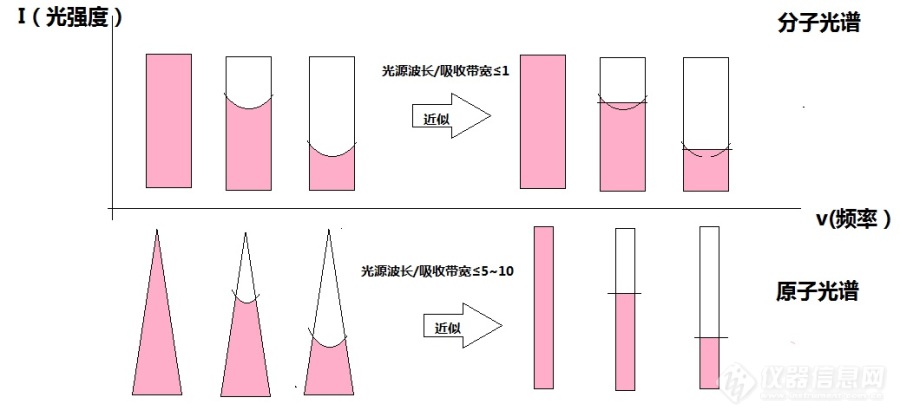
[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url]中经常提到,因为空心阴极灯的光谱很窄(0.002nm),计算积分吸收系数不易,故以峰值吸收值代替积分吸收。但实际根据Lambert-Beer定律,实际测定的是光的强度I0和I,这本身就是积分吸收值啊,得到的a相当于平均吸收系数,和峰值吸收有什么关系呢?[img=,690,320]http://ng1.17img.cn/bbsfiles/images/2018/05/201805162237123439_3468_1340227_3.jpg!w690x320.jpg[/img]
液相色谱开发时,对物质进行用紫外扫描确定最大吸收波长,溶剂是要用流动相还是什么都行?
我们使用的分光光度计是最简单的那种,无扫描装置。如果用标准溶液测定最大吸收波长,是否可以这样测定:选用石英比色皿在紫外区域测定不同波长下的吸光度,再选用玻璃比色皿在可见光区测定不同波长下的吸光度。从中选出最大吸收波长。
我有一样品,在365nm波长下连续照射的情况下,会不断产生一种新的物质,这种新物质可以在400nm和600nm左右有吸收,因此,可以在照射后每隔50秒扫描一次,这样在400nm和600nm左右处会有吸收峰,而且,随着照射的时间增长,峰会越来越高,即峰按50s、100s、150s、200s的顺序增大。可以在普通的紫外可见扫描仪中做这个实验吗?我今天试了,没试处来,用的紫外比较老,岛津UV-1601,里面有指定波长,而且有重复扫描选项,可是指定波长365nm,每隔50s扫一次,每次得到的图都一样(当然可能是溶液没配好)。现在的问题是用紫外扫描仪可以做这种固定波长,并连续照射,然后每隔一定时间扫描一次样品,得到吸收谱图吗?是不是高档的紫外扫描仪才能做?或者我的照射实验应该单独在外面做(紫外仪中的强度不够,不足以激发产生新物质),然后每隔50s取一次样品,测它的吸收光谱?只是这样做时间上不好把握,误差很大。请大家指教,如何做好这样的实验?
看了一篇文章,做植物内生菌分离之后,再筛选可产生PQQ(吡咯并喹啉醌)的内生菌的步骤如下初筛:选择加入甲醇后发酵液变为红色的内生菌(说明选择是甲醇利用型内生菌)复筛:原文如下”PQQ 的光谱法分析参照杨延新的方法进行,将 PQQ 产物分别加入石英和玻璃比色杯中进行紫外扫描,以 MPQ 培养基加入 3 倍体积甲醇液为对照,记录 D326nm和 D400nm值,计算OD326nm-OD400nm值用以检测 PQQ 含量“不明白之处是为何减去400nm处的值。随后查杨延新的文献上又有如下解释“光谱法是以 D326nm值与 D400nm值的差值来检测PQQ 含量。 PQQ 在 247 和 330 nm 左右有 2 个特征吸收峰。考虑到蛋白质和核酸的干扰,排除 247 nm的吸收峰,选择 330 nm 吸收峰的峰值吸收,即 326nm。 400 nm 为末端吸收,利用 326 与 400 nm 的差值可以消除系统误差。”不明白到底什么是末端吸收,查了相关解释也是一懂半懂,400nm处究竟什么引起系统误差??后来问过别人,说是因为如果做全波长扫描的话,在400处有吸收峰,400处值不是最低,所以要减去400的值。可是明明是326是PQQ的最大吸收峰,而要测得也只是PQQ而已,到底为何减去400的值,好纠结,不懂啊~~~~~对紫外吸收这块实在很初学,麻烦高手解释一下~~~~谢谢~~~
关于[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光光度计检定规程的几个问题:1、静态基线稳定性,光谱带宽0.2nm,量程扩展10倍,点亮铜灯,在原子化器未工作的状态下,按以下操作:(1)单光束仪器与铜灯同时预热3分钟,有“瞬时”测量方式,或时间常数不大于0.5秒,测定324.7nm谱线的稳定性,即为30分钟最大漂移量和噪声(峰—峰值)。量程扩展10倍是什么意思?。2、背景校正能力检定:对于带石墨炉的集仪器,将仪器参数调到石墨炉法测镉的最佳状态,以峰高测量方式先进行无背景校正测量,用移液管加一定量的氯化钠溶液(该溶液浓度为5.0mg/ml,必要时可稀释),使产生1A左右的吸收信号,读下吸光度A1,再用有背景校正方式测量,加入相同量的氯化钠溶液并记下吸光度A2。试问“1A左右的吸收信号”的“1A”是指多大的值?
求教大家,再石英片上测薄膜吸收,膜厚微米量级,以空白石英片扫描基线,发现薄膜的吸收和基线几乎重合,没有峰后又增加膜厚,采用滴涂后固化,吸收有所增加,增加的大概0.5左右,但是几乎没有峰值
[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]法的基本原理中的积分吸收和峰值吸收还是不太明白积分吸收稍微有点明白了,但是峰值吸收又是什么意思呢?峰值吸收为啥要用锐线光源呢?最终实际应用的时候,测定的是吸光度,吸光度和单位体积内被测元素基态原子数有关系的,这又和峰值吸收没关系了吧?哪位大侠能给小弟详细解释一下
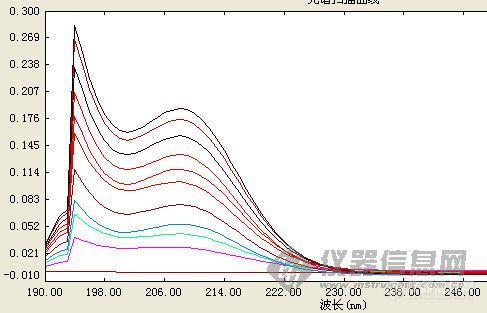
紫外可见分光光度计为普析TU-1810PC测量的物质为二甲亚砜图1的溶剂为去离子水图2的溶剂为甘露醇溶液(加入甘露醇的原因是考虑到-OH是个助色团,自己也是抱着试试看的心情测的)图1的结果之前发帖问过,很多人说可能是由于波长200nm,仪器的性能有限所致。但图2同样也出现在峰的后半段出现几乎直线下降,但此时的波长还是大于200nm的啊。另外图1有两个吸收峰,而图2只有一个吸收峰,是因为溶剂不同的结果吗?达人指教,谢谢![img]http://ng1.17img.cn/bbsfiles/images/2009/10/200910181906_176319_1813953_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2009/10/200910181907_176320_1813953_3.jpg[/img]

合成了一种材料,带氨基,加入苯胺蓝后颜色变得很浅,肉眼几乎无色,但是直接紫外扫描紫外区吸收峰太强,将其与苯胺蓝原液一起稀释50倍后扫描光谱图如下:发现紫外区吸收峰增强,可见区吸收峰减弱且蓝移,染料是被吸附了?还是发生化学反应了?如果是吸附不应该是整个光谱区吸光度整体下降么?http://ng1.17img.cn/bbsfiles/images/2015/12/201512281138_579683_2980891_3.jpg
我们用PE的紫外分光光度记做样品浓度的测试,用的是扫描,平时测的时候最好吸收峰都在713nm,以此我们来观察样品的浓度的,可这段时间最高吸收峰却移到723nm,再连续做几次又会移回713nm,请问各位朋友这是为什么呢?
请大家帮忙, 在使用[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]时,打开[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url],在仪器初始化时,放生嗤嗤的响,灯电流和狭缝已初时化完成,显示ok,扫描波长时发出的声音,重开几次机时,就没事了,请问是什么原因?应该怎样调整?
请问大家一个非常低级的问题,不要笑话啊。[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱仪[/color][/url]和[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光光度计是一个概念吗?如果不是,请问有什么区别呢?







 我要推广仪器
我要推广仪器
 下载APP
下载APP