手动调节氘灯电流,是否可以调节基底复杂的样品的背景氘灯由于容易背景扣除过度,是否可以通过手动来调节氘灯电流来调节扣背景?
请教诸位一个问题,现有液相色谱制备型手动进样阀有没有定量环超过20ml的?如果有,能达到多少?还有液相色谱糖柱能检测多糖吗?分子量达到多少的?
手动积分如何降低背景噪音影响。
本人最近在做毕业论文实验(大四) 课题是顶空法测定葡萄酒中的甲醇由于条件限制,只能手动进样,就是水浴箱加热,进样针手动进样仪器岛津GC-2010 顶空条件 70度 恒温 30分钟色谱条件 进样口200度 分流比10:1 柱流量 1.50ml/min柱温 45度4MIN 后5度/MIN到70度FID检测器 温度:220度色谱柱是SUPELCOWAX-10 (30M*0.32mm*0.25um)由于条件限制 只有100ML刻度的顶空瓶 加样量50ML 进样量 1ML问题是:1.对照样中内标(正丁醇)和甲醇的浓度相等(约300mg/L ),可是出峰后甲醇的峰面积很小,大概只有内标面积的0.2~0.4倍,为什么? 2.我用的是震荡水浴锅,但由于水浴锅在8楼,气相在5楼,这来回会不会影响平衡?恳求前辈们帮忙,现在只求能使出峰面积规律一点,不要时大时小,能做一个差不多的标准曲线就能完成论文了。。
液相手动进样时,进样量一般为定量环的几倍时,定量比较准确啊?谢谢啊
请教大家,请问topas4.2可以手动编辑背景吗(有时候觉得函数拟合的就是不太满意)?非常感谢!
我看到有些人测试核磁基线不平的时候,手动调节基线就会好很多,谱图也会很漂亮,我知道积分是和每个人的习惯做法有关的,但是都会尽量客观,但是如果手动调节以后还会准确客观么?谢谢!
同志们好,最近想买个手动薄层涂布器,自己铺板子用,但是一直找不到卖的地方,谁知道在哪儿可以买到啊,知道卖家的联系方式也好啊,着急用,谢谢各位!!
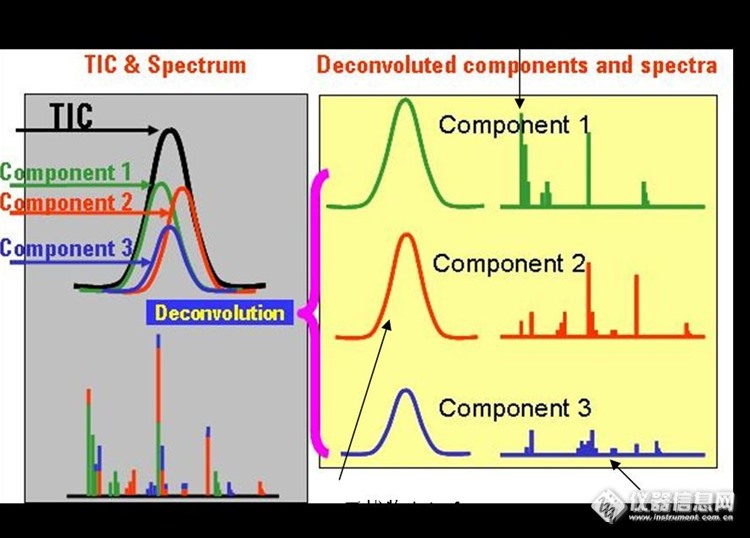
AMDIS自动化质谱图解卷积和鉴定软件在GC/MS数据处理的初步应用(6)- 手动背景扣除 先回顾一下AMDIS的基本概念对于AMDIS有的网友可能比较熟悉,特别是农残,环境,有害物,香精香料等领域的朋友可能属于高级使用者。本人以初学者的身份初步介绍一下AMDIS。如有不妥,请批评指正。未经许可,不得转载,请谅解。一般来说,目标化合物的分析要求检测目标离子和确认离子的比例。然而,对于高基体背景的样品,大峰后面的痕量组分或流出时间很接近的成分,离子比例会受到基体的影响很难符合要求。为了确保分析结果可靠,一般采用背景扣除及手动积分。因此,对于复杂基体的样品数据处理,需要耗费大量的时间。为了提高分析效率,谱图可以利用一种称为“解卷积”的数学计算来将目标化合物从背景中分离出来。美国国家标准和技术院(NIST)开发了功能强大的解卷积软件,即自动质谱解卷积和鉴定系统(AMDIS)。下面简单介绍一下AMDIS:AMDIS软件由美国国家标准技术研究院(NIST)(National Institute ofStandards and Technology)提供。The Automatic Mass Spectral Deconvolutionand Identification System (AMDIS)自动质谱图解卷积和鉴定系统软件(AMDIS)让您从GC/MS数据文件自动找到目标化合物。软件先对GC/MS数据文件解卷积寻找所有分离组分。每一组分与目标化合物的谱库进行对比。如果以上的用户设定值,然后报告出目标图谱和解卷了组分的图谱的匹配因子。什么是解卷积(Deconvolution)?NIST AMDIS的定义:“这里所用的术语在广义上是指从一个复杂的混合物中提取信号。 解卷积的过程包括处理噪音、校正漂移、从紧密相邻的共洗脱峰中提取出单个峰等。” (简单讲就是去复杂化)用下面的简图可以解释解卷积过程:在GC/MS 中,Deconvolution是一种数学技术,它可以将重叠的质谱图“分开”成为“清晰”的单个组分的谱图。图1 是这个过程的简单示意图。这里分别是总离子流色谱图(TIC)和质谱图。与常见的情况一样,这个色谱峰包含了多个重叠在一起的组分,而最高点质谱图实际上也是这些组分的组合图。质谱谱库检索只可能给出一个较差的匹配,而且不能识别所有构成这种组合谱图的单个化合物组分。http://ng1.17img.cn/bbsfiles/images/2016/12/201612152312_01_1615838_3.jpg图1 解卷积过程的简单示意图其它相关内容请参考我以前的帖子。
有谁做过顶空气象色谱法手动进样测水中的氯乙烯的吗?我用的是安捷伦7820A,HP-5柱子。出来一个平而缓保留时间有近3分钟的突起,这是怎么回事呢?谢谢

http://ng1.17img.cn/bbsfiles/images/2011/07/201107142307_304968_2088866_3.jpg安捷伦 5890-5973 GC-MS.我手动进样六次,然后第一个峰是含量测定用的。 有点过载了。我没改分流比。求帮,这样算合格了吗?
在测试邻苯二甲酸盐,听别人说DIDP和DNOP需要手动积分,但教材上根本没提到如何手动积分啊@还有就是如何扣除背景?请哪位仁兄指点一下。谢谢!
手动调谐输入丙酮58峰,可为啥执行轮廓图时显示的峰值是却是62.1?
做了三年多的进口仪器PE-800,日立-2000全是自动进样,最近接手一台普析通用的990[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原吸[/color][/url],还不带自动进样器,很无奈……手动进样出现的问题是,样品推出后黏在枪头上,移开枪时便会堵住进样口,希望有经验的前辈传授点经验,谢谢了
由于一些原因,最近做样出的谱图总是不太好,标样和样品的谱图放大后看不一样,致使峰面积不一样,得出的结果不如人意!于是乎手动积分调整了下,这样结果虽然很好,但心里又有不踏实的感觉!系统自动积分是按一定规律,手动调整就不一样了,想跟大家讨论下这个问题!畅所欲言吧!
目前很多制药企业都主要以自动进样为主,只有学校和一些中小型的第一方实验室使用手动进样。小编最早做实验用的就是手动进样,实不相瞒,测盲样儿测得简直要吐血,重复性太差,一遍一遍的测。。。说白了,手动进样考的就是个熟练度,熟能生巧,再加上有经验了自然数据就可信度高,重复性那还叫事儿吗。说起来,除了熟练基本功,还有哪些经验型的东西值得总结呢?重复性差原因手动进样导致的面积重复性差原因主要有以下:1、进样量不合适;2、所用进样针刻度不准;3、进样针里有起泡;4、进样口松动,针插不紧;5、进样针太细,插不紧;6、转子密封垫脏、磨损或漏液;7、进样方法不正确。可以自己对照逐个排除。进样量的把握以20ul原配样品环为例,部分注入样品进样体积在5-15ul重现性好,建议10ul。全部注入样品进样体积以20*4,即80ul重现性好,就是要达到样品环体积4倍以上。手动进样步骤1 吸样后擦针、排气泡;2 进样状态(inject)下插针到底部;3 快速切到装填(load)状态;4 较快匀速推针杆,注入样品;5 快速切回inject;6 拔针。步骤要牢记,速度要把握好,熟练才是最好的。
这是我上次日本电子工程师来清洗电镜的时候传授的一点心得。因为一直很忙,就要过春节了,所以赶紧写一点来一起分享手动调节Gun Alignment到最佳的一般步骤。机子型号:日本电子6063LV在换灯丝或者是物镜光阑后一般都要手动调节Gun Alignment。特别是要做能谱的这点显得更加重要。虽然很多仪器都有AUTO调节的。但我个人认为手动的可能效果更好,而且掌握一下如何调节总没有错的。1。在放大倍数到最小状态,适时可以变化,spot size 调节到30,tilt X、Y调至中央。shiftX、Y也调节到中央开始。2。调节LC从左到右,直到黄带位置。(此过程会发现视图由暗变亮,后又变暗,再变亮。此sean wen曾经贴过图[img]http://ng1.17img.cn/bbsfiles/images/2006/01/200601181810_13273_1608025_3.jpg[/img]能很好的说明。我们就要找这前面的那个饱和点。3。AUTO ACB,对比度好了,倍率调节到最低。4。分别调节tilt X、Y直到视图都最亮,中间可以再次AUTO ACB。5。分别调节shift X、Y直到视图都最亮,中间可以再次AUTO ACB。6。调节LC到最亮,如果比较不出来就要AUTO ACB(调节对比度)7。再调节tiltX、Y和shiftX、Y。使视图最亮。8。增大倍率,调焦,AUTO ACB *10000。10。调节OL WOBBLE。再调节倍率最小。调节tiltX、Y 使视图最亮。11。增大spotsize,应一直变亮,如果看不出变化现象,应再次AUTO ACB后再调节。若在增大spotsize的过程中,有变暗的情况时,调节shiftX、Y。使视图最亮,直到调节spotsize达到90左右时一直是视图在增亮的过程。12。把spotsize调到30,再调节tiltX、Y。使视图最亮。13。倍率增大,调焦后OL wobble使视图不在晃动。14。变小倍率,增大spotsize检查是否有变暗的情况。如果有重复11步骤。15。spotsize调回到30,再次调节tiltX、Y。使视图最亮。16。放大倍率,调焦,调节stigmX、Y、OL wobble。17。观察LC电流,一般有这样的关系:30KV在80+/-5uA、20KV在70+/-5uA、15KV在65uA左右。如果LC电流不在此范围内,可以点击set bias,调节coarse大小来调节LC电流。注意他们之间的关系。18。在换灯丝的第二天,LC最好回调一点(这点其实很重要,可以延长灯丝的寿命)大家想想为什么。
除统一使用工作站默认积分参数外的所有参数修改都属于手动积分吗,如修改斜率、最小峰面积、积分开始与结束等,这些修改也会造成基线的微波调整?以下是一些网络摘录的理解:[quote]关于手动积分的检查不合格项(转载自丁香园:对手动积分的认知)[/quote]在2014年,FDA对欧洲某实验室的检查中,483表格中有一个观察项这样表述:没有描述如何进行手动积分的操作规程(No procedure exists describing how to perform manual integration)。在2007年8月,FDA给Leiner Health Products公司的警告信中,有一条缺陷项这样表述:检查员记录了很多“数据捏造(manipulation of data),却没有解释捏造原因”。这个捏造包括:改变色谱积分参数、重新标记峰,这样之前本应该积分的峰就不再积分,不计入杂质的计算。这说明了FDA是接受手动积分的,但应该有操作规程控制手动积分的条件、权限、注意事项等,不能背离科学地通过手动积分使检验结果合格。备注:在FDA的现场检查中,例如发现预进样、重复检验的情况,检查员的观点是:如果企业证明是科学的、必要的,就应该写入操作规程。这也意味着,需要经过充分验证、科学评估。[quote]手动积分的担忧[/quote][align=left][img=,682,359]https://www.ouryao.com/data/attachment/forum/201512/15/093153nwql6wxd3slfqqz1.png.thumb.jpg[/img][/align][quote]手动积分的用处[/quote]理想条件下,在通过系统适用性试验后,标准品、样品的色谱应一直以相同的方法进行积分。但是实际情况并非如此,尤其是接近定量限、检测限的情况下,有些峰自动积不上,需要手动积分。手动积分虽然会受主观因素的影响,在科学分析色谱结果时也是需要的。举两个例子说明:例1,当出现异常峰,分离度不好的峰时,可以手动强制分割。例2,研发阶段,API、稳定性样品的纯度、降解产物的检验方法色谱运行条件相同,但应考虑到检测分析的重点不同,可能需要不同的积分参数。在这种情况下,最好建立不同的分析方法、程序。[quote]关于手动积分的指南[/quote]关于手动积分,并没有指南给出明确的定义。笔者只查到有两篇生物样本分析的方法验证指南规定了再积分(Reintegration)的注意事项。[color=#555555][/color]FDA的2001年定稿指南 Bioanalytical Method Validation,及相应的2013年草案指南也有类似的要求。 生物样本分析的方法验证2013草案指南,第III.C节“样本数据再积分”规定:SOP应该确定再积分的标准,和怎样进行再积分;应清楚描述、并记录再积分的理由;应报告原始和再积分数据。第VII.D节还规定:再积分数据应有管理人员授权再积分。EMA的2011年指南 Guideline on bioanalytical method validation,第5.5节规定:应该有SOP描述色谱的积分和再积分。应在分析报告中讨论偏离这个SOP的任何偏差。应该记录色谱积分、再积分的积分参数、初始积分数据、最终积分数据,并在要求时立即可得。[quote]没有公认的手动积分定义[/quote]目前没有关于手动积分的公认的定义,Hill的一篇文章,讨论了手动再积分的范围和术语,并总结认为,建立一致的定义是有必要的。如果没有手动积分的定义,企业怎么建立积分的SOP呢?[color=#555555][/color]在2015年11月,美国费城的ISPE年会上,企业人员Bob McDowall和Mark Newton建议区分手动干预(Manual Intervention),和手动积分(Manual Integration),并提出:手动干预 :没有手动改变基线,例如:1)对峰重命名,或调整积分窗口;2)改变积分参数;手动积分 :分析员手动重新划基线。举两个例子说明:例3,在色谱峰的保留时间不在预期的时间窗口内,需要手动干预时间窗口,而峰面积不会改变;需要调查保留时间偏离的原因。可能FDA会把这个干预也归为手动积分;但是Bob认为不必归类为手动积分。所以希望监管机构给出明确定义的指南。例4,有IPEM学员单位提议:手动干预、手动积分这两种方式在有些情况下可替代使用的。峰无法识别(或不能正确识别)时可能通过手动积分(重画基线)或调整积分参数解决;峰积分不好时可通过手动积分(重画基线)或调整积分参数解决。[quote]手动积分使用的注意事项[/quote]通常,在得到自动积分色谱图后,分析保留时间、峰型是否如预期,峰是否被正确识别,基线是否满足要求,或者其它标准。只有在色谱图不可接受时,才可考虑是否可以进行手动积分,或者进行实验室调查。[color=#555555][/color]有两种情况,是禁止进行手动积分的:[list][*][align=left]在自动积分后,对称的峰有可接受的基线;[/align][*][align=left]增强峰、或削减峰的目的是为了满足系统适用性接受标准,或满足放行质量标准。[/align][/list]所以,企业应建立科学、合理、透明的自动积分操作规程,并满足数据完整性的要求。在SOP之外进行的手动积分可能被视为数据造假。[quote]色谱软件选型、权限设置和结果显示[/quote]满足合规要求的色谱软件(例如Waters和Angilent的软件),能够分别设置不同权限,例如普通化验员不具备进行手动积分的权限,需要主管进行操作,有些软件则不具备这些功能;即使是同一个品牌的软件,也有不同版本;即使软件有这个功能,企业也不一定启用。 (企业已经在用的老系统legacy system,无法通过软件权限控制的,只能通过SOP进行控制,同时建议基于风险逐步替换为新的完全符合[url=https://www.ouryao.com/]GMP[/url]的系统)[color=#555555][/color]有两个信息是否显示,对色谱数据透明度非常重要:方法名称和数据版本。 如果企业不使用手动积分(重画基线),而采用修改参数的方式,则每修改一次,是不同的方法(规范的管理程序会要求更改方法名称保存,否则就只能到audit trial才能查出来这个方法已经被修改了),而打印的色谱图可以选择是否显示方法; 每处理一次数据,会形成一个数据版本,同样的,打印的色谱图可以选择是否显示数据版本。一个完全透明的结果显示,会在打印的色谱图上显示“方法名称+数据版本” (同时要求修改参数则改变方法名称,不需要QA或检查员仔细检查audit trial才能发现修改痕迹)。[quote]QA产品放行时对手动积分的判断原则[/quote]“手动积分应以能获得更准确的检测结果,并且不增加产品质量风险为原则,同时及时记录相关的科学依据。”[color=#555555][/color]以文章第2小节“手动积分的担忧”中的色谱积分为例,图中哪种操作是放大质量风险呢?答案是视具体情况而定。 例如如果左边这个峰是主物质的峰,这个检测是含量(或纯度)检测,则“ 红线(左),削减峰的积分方式会造成峰面积的偏小”实际上控制得更严,如果我们知道在这个位置可能存在一个其它杂质,则这也许是一种适当的手动积分处理;反过来,如果左边这个峰不是主物质峰,它是一个杂质峰,其意义正好相反。 如果这两个峰一个是主物质峰,另外一个是杂质峰;而另外一种情况,这两个峰是这个药里面的两个组分,分别有不同的含量标准;同一个手动积分操作的意义,对产品质量风险的影响是完全不同的。这里有太多可能性,在这里不详细描述每一种情况了,但其原则(基于科学,力求准确,不增加质量风险)是明确不变的。[i]感谢IPEM2007级学员闭欢欢及单位同事邓玉庄、林宾、黄仲仪对本文的贡献。[/i]作者:识林-榕
借着有网友讨论自动进样器的事情,我来讲一讲手动进样器的故事:石墨炉离不开自动进样器,性能再好的石墨炉,没有自动进样器配合很难保证精度,也使分析变得十分乏味。只有一种情况除外:我的好友在做地质化探样品时,嫌自动进样器太慢,熟悉的操作足以满足分析的需要,而用手动进样快得多。现在的自动进样器已经令那些早年从事[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分析者感慨不已:自动浓缩,自动稀释,灵活加入多种基体改进剂,标准加入法和标准回收测定,还能自动进行质量控制,用一个溶液制作校正曲线等等。各种功能,应有尽有,令人眼花缭乱。我们称其为:已经满足了现代石墨炉分析的需求。据了解国内也有几个厂家做出了很好的自动进样器,除了瑞利的用于石墨炉外,吉天的还用到了氢化物发生上,不知以后能不能于石墨炉联用做在线富集。想想在早年,一个eppendoff的手动进样器还要1000多元,那时候真是太奢侈了。许多单位还拿医用的进样针来进样,玻璃的外壳,一根不锈钢推杆,靠刻度控制进样量,那个细细的针尖插到石墨管里可不是每个人都能干的。我曾经看到许多小女孩(现在也快变“老太婆“了)用那玩意儿做出极高重复精度的分析结果来,除了赞叹之外,还只能战战兢兢,绝不敢贸然与他们比试!记得“西北狼”有一次也在网上和大家讨论手动进样,我插了一句,说:学会手动进样还是很需要的。我已经查不到发在哪里了,[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]板块人气较旺,几天前的帖子就可能很难找到,幸喜大水牛现在做了个相同主题的链接,虽然还很不完善。不过,说还需要手动进样,这话一点也不夸张。下面讲的就是我的真实故事:1.氯化镁对铅原子化的干扰:还是在80年代,我和一位同仁用石墨炉做滑石粉里的铅,那时候还没有多少基体改进剂的概念,只知道可以用标准加入法,所用的自动进样器也还不能自动做标准加入法。曲线做得很好,做样品几乎没有信号。翻来覆去作结果信号越来越没有规律,连标准曲线也做不出来了。不知怎的,重配标准又能作出曲线来了。闹了整整一天,没结果也没头绪。再稀释一次标准溶液,样品杯等也清洗的干干净净!再来一次。我说:也不要做标准曲线了,我们直接拿一个标准和样品比较比较看。那就拿来个手动进样器直接进样得了。吸了一个标准,嗯,测得很好。吸一个样品,没什么信号。再吸原来那个标准,呀,见鬼了,没信号了,再吸标准,怎么也做不出信号了。只有一种可能!我想起来了,那时候,研究铅原子化[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]干扰最重要的例子就是氯化镁对铅的干扰!滑石主要成分有碳酸镁,经过处理还有很多氯化镁残存。一定是它,难道有那么严重?那就再来一次,再配标准,好!标准又做出来了!作样品,没信号,换个进样嘴再做标准,又能出峰了。再做样品,不换进样嘴,做标准,没信号了!哈哈,这下现象重现了!就那么一点样品的接触,那个进样嘴也是eppendoff的,厌水性很好的!太夸张了,可又那么现实!样品中加一点抗坏血酸,好多了。虽然还没有更本解决,总算是找到问题了。我现在回想起来:如果我们就是使用自动进样器,一定很难发现这个现象的,就是发现这个现象,要去重复它,恐怕用自动进样器比手动进样更烦。我猜想。2.铅的容器污染:某研究所做中药中的铅的石墨炉测定,他们也是刚开始做,经验不是很多。一天打电话告诉我:标准曲线做不出来,没有相关关系,有时标准空白吸光度很高,比后面的标准还高。好吧,我说,我去看看。到了那里我一看:配标准使用的都是玻璃的。我说,你看你怎能用玻璃容器配标准呢?玻璃容器还不一样,那个带青色的瓶子是铅玻璃,那个带点黄色的是钠玻璃,这样怎么能做出标准曲线来呢?那怎么办?他问。我说:我们试试,拿了一支手动进样器,取一个样品杯里的溶液,第二针进样的体积等于第一针的两倍。结果测得的吸光度很明显与体积呈正比关系。我说:你看,肯定能做好的,只要你把容器换成聚四氟乙烯的。你不要老守着你的自动进样器,做一些实验的时候,手动进样是很方便的。第二天,他打电话来说:没问题了,其他条件再摸一下就可以了。我还是在想:会用手动进样还是能更灵活的试验条件观察各种现象的。现在石墨炉分析时,很多仪器一接上自动进样器,就是没有你什么更改条件的余地了,一切按照它的死板的程序继续。实际上是一件很不愉快的事,特别是在摸方法条件时。打个比方,在分析前想看看石墨管里有没有残留物,它就不允许,好像每一个测定都是必然成功的似的,所有数据也都被记录下来。我要是编写石墨炉自动进样程序的话,一定要设一个可以不进样(用自动进样器)就执行加热并可以观察的程序。3.饱和食盐水中钙的测定:离子膜制碱对于进入池中的氯化钠溶液又很高的要求,记得好像钙镁的合量不能超过20ppb。这样低的浓度通常还使用石墨炉测定,(不过现在有用ICP-OES成功测定的典范了)。做这样的分析石墨管必须用高纯的,那是用氟利昂净化过的非热解涂层石墨管。另外,水,环境(空气中的粉尘)太重要了。用亚沸水,从这个实验室走到另一个实验室就不行了。实际上,氯化钠对钙信号的抑制很强,我们制做的亚沸水在石墨炉里测钙条件下都能得到0。3吸光度(条件不太好),加了样品就几乎低十倍。但是钙镁石墨炉灵敏度是太高了,足以弥补。这样的话只能做标准加入法,说实话,要配2ng/ml的钙标准,我还没见谁有那么好的条件把它配出来过。只有在饱和食盐水中直接加入小体积高浓度的标准溶液,例如:对100毫升的饱和食盐水加入20微升10ug/ml的钙标准,就等于溶液中加了2ng/ml的浓度。这下手动微量进样器又有用武之地了。你信不信:但我们用手动进样时,得到的测定精度还比用自动进样器好(那时的自动进样器确实不如现在的好),原因是,自动进样器加了几个样,食盐水就粘在进样毛细管上了!讲了几个故事,都是用手动进样作石墨炉分析的。一来让我们的年轻人了解一下“老人“们在那时是怎样干活的,二来还想说明,掌握手动进样技巧可能还是有用的,特别是在摸索条件,观察干扰等场合;也可能在污染严重需要减少使用的器皿,或者一些物理性质特殊的样品时,它也会有用的。Sorry!现在说这些,可能不合时宜了吧!只算一个故事......
大家好,做农药残留,手动进样,想用外标法,如何判断我出来的谱图是准确的呢?具体是怎么样操作啊?是不是只要每个样品多进几针,然后比较谱图,有大概3针左右的目标峰的峰面积差不多高就可以了啊?手动进样时,总觉得外表法不是很准确,同一样样品进去的,峰面积会有高低。。
今天在电子天平说明书上看到可以选择正常称量和手动填充称量两种模式,不知手动填充称量是什么意思,望各位前辈指点一下[em06]
各位高手,我是个新手,想请教一下液相谱图中怎么手动计算噪声和信噪比,软件上有这功能,但就想了解一下自己算的方法……
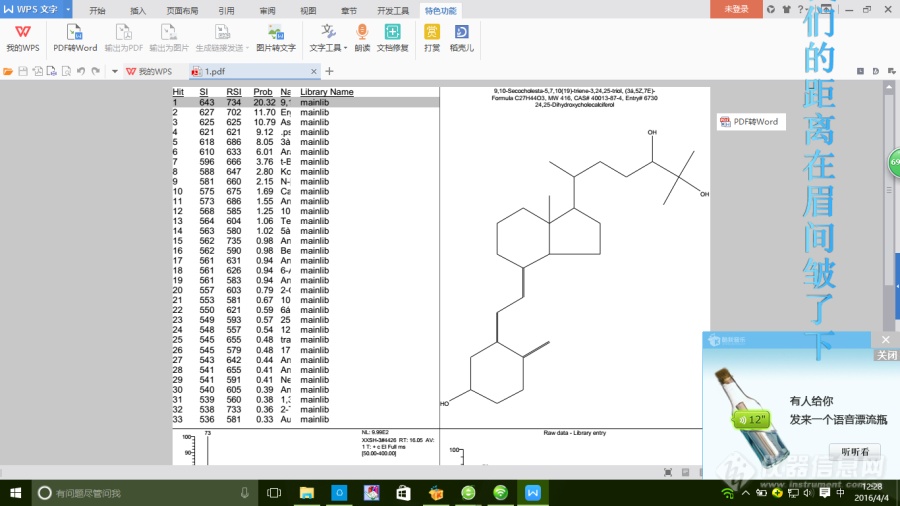
我想问一下质谱手动导出来的谱图要怎么分析啊?根据什么来定量?质荷比是要看什么的?[img=,690,387]https://ng1.17img.cn/bbsfiles/images/2019/08/201908051348198578_905_1853141_3.png!w690x387.jpg[/img]
有没有哪位前辈了解仪器分析中线性方程和线性系数方面的知识,在ICP分析中,仪器会自动生成线性系数,但如果是手动计算,该怎样计算?
谱图出来后,各个组分的峰 面积自动被切出来叫自动积分,不知道我的理解是否正确?另外通过手动来改变切出来组分 的面积是不是就是手动积分呢?如不是请指教什么是自动积分,什么是手动积分。另外手动积分需要改变哪些参数?谢谢高手指教。
最近做顶空,不小心将手动进样针摔碎了,杯具啊,进样针为什么都是玻璃材质的呢?有木有不怕摔的进样针呢?
现在大家都习惯了自动化,自动进样,全自动电子天平,自动定量计算等。请问您还会手动进样,手动作一下事情吗?
在论坛潜水多日, 默默和大家学习了很多。但是作为一名小白,还要很多不懂的地方,我看论坛经有经验的前辈经常提到通过调节Z0, lockpower, lockgain, lockphase手动锁场。有谁可以分享一下,怎么手动锁场吗?详细的步骤是什么?我这边是bruker AVNEO 400M.谢谢大家
ZESSI 50电镜,现在想做钢铁材料高倍20000X左右的扫描组织中析出相观察,但是电镜象散不好怎样调节象散使其最佳?谢谢![em09502]我以前调过一次,是这样顺序灯丝像对中,放大到10000倍,手动调象散,调好后,微调focus wobble。
[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]手动积分怎么积分误差才会小点,每次手动积分的结果都不一样,手动积分有没有一个统一的标准?







 我要推广仪器
我要推广仪器
 下载APP
下载APP