各位大神最近咨询了好多单位的老师,大部分老师说全自动闭口闪点准确度较差而且还容易出检测事故,手动闪点准确度比较高是吗?
安捷伦7890-5975气质联用如何手动删除积分 手动积分可以有 但是没有找到哪里能手动删除
有谁做过顶空气象色谱法手动进样测水中的氯乙烯的吗?我用的是安捷伦7820A,HP-5柱子。出来一个平而缓保留时间有近3分钟的突起,这是怎么回事呢?谢谢
GC工作站LabSolutions工作站图谱手动积分后打开报告模板左上角出现一串字符,有点像页眉。但删不掉,如果不手动积分,直接打印谱图就不会出现。版本2000-2004[img]https://ng1.17img.cn/bbsfiles/images/2018/12/201812051239001431_3168_3508821_3.png[/img]
请问大家做滴定分析还是用手动的滴定管滴定吗
样品:测定氨水浓度(氨水经过充氨处理,应在28%左右)测定方法: 酸碱滴定 1.手动滴定 取2ml氨水,加30-40ml稀释水,用甲基橙作指示剂,12.6mol/L 盐 酸滴定,消耗体积平均为28mL. 2.电位滴定 取2ml氨水,加约50ml稀释水,用复合电极12.6mol/L盐 酸滴定,消耗体积平均25.6mL.问题: 两种测试方法,各自的平行性很好,为何两者偏差太大原因?为什么电位滴定结果偏低? (又及电位滴定时也加入甲基橙,也在仪器判别终点处变色,体积误差很小)
各位大侠好,我是做水质监测的新手,没见过顶空进样器,现在我们是采用手动式顶空进样。在试验中出现这样的问题,在做水中吡啶的测定时,先做的标准曲线,线性达到0.999,接着处理了一批样品(规定:在水浴锅中平衡30min),进样发现峰面积较理论值偏高50%,试样进样结束以后,又进了先前的标样,标样的峰面积也有相应的升高(标样也同时放置在水浴中)。我的问题是:1、在顶空进样中,样品的平衡时间怎么控制,涉及到大批量操作时,怎么保证每个样品进样前刚好平衡了标准规定的时间?2、样品的平衡时间的长短对峰面积有什么影响?我原来的理解是应该类似于趋于一定时间后,峰面积就基本保持不变了,实验结果是这样的情况,是因为大批量操作,导致平均每个样品的平衡及进样时间较长,峰面积大大增加吗?3、在手动进行顶空进样时,有什么注意事项,怎么保证样品处于动态平衡状态?在手动进样时,峰面积时大时小,是样品处于不平衡状态?还是进样的手法问题?请各位大侠指点,可能问题问的有点外行。详细讲解下顶空进样器的大批量操作相关细节,或手动顶空进样的一些注意事项,传授下经验。谢谢了
[color=#444444]实验室需要用到顶空[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url],但是订的自动顶空进样器还需要比较长的时间才能到,因此需要先做顶空的手动进样[/color][color=#444444]目前用的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]是安捷伦的6890N,现在要做的是定量分析,用的内标-标准曲线法,进样是用1 ml的一次性塑料注射器,效果一般,线性还没符合要求[/color][color=#444444]因此想请教各位大神,如果要做手动顶空进样的话,进样针的选择有什么注意的或者有什么推荐的吗?[/color][color=#444444]感谢各位的指导[/color]
用顶空进样与手动进样结果有什么不同?为什么手动进样有时分不开
适用标准:GB/T3536-91(克力夫兰开口杯法)GB/T267-88 产品说明:本仪器采用当代先进技术,集机械、光学,电子及计算机技术于一体,结构紧凑。可自动完成石油产品开口闪点的测试,实验过程中的升温速率及扫描点火动作由微机控制自动进行,油气闪火时,检测系统自动捕捉并记录闪点温度。对闪点数据进行自动气压修正,并以规范的格式打印输出,还可根据需要重复打印数据。本仪器可由计算机监控(无线/有线通讯方式,由用户选配)。本仪器结构合理,性能稳定,操作简单,是理想的分析检测设备。主要技术指标:1、测温范围:-40℃-400℃ 分辨率:0.1℃2、测温原件:PT100(德国JUMO公司原装测温传感器)3、闪光检测:采用国际最先进的光学检测系统4、加热方式:金属浴加热,微机双路闭环控温5、点火方式:气体火焰点火,电子自动引火6、自动灭火:闪点出现后自动执行灭火动作7、控制臂自动升降产品特点:1、通过控制油样温度及加热器温度可实现升温速度的均匀稳定;2、德国JUMO公司原装PT100测温传感器,确保实验数据与标准手动实验数据相同;3、自动灭火装置确保实验安全。备注:本仪器采用大屏幕汉字显示,操作直观。本仪器可通过快速启动的方式启动试验,操作简便。本仪器可预先设定8个控制程序,满足对不同油品的试验。每一次扫描点火前,都执行一次电子引火试验,确保火焰不熄灭。仪器内置气体控制电磁阀,试验结束自动关闭,确保安全。本仪器可按GB/T3536-91(克力夫兰开口杯法)或GB/T267进行试验,试验方法选择在相应的控制程序中设定。打印机可通过软件控制其开/关,关闭打印时,只显示试验结果。配备内部时钟,无需输入实验日期有效使用年限95年。
手动进样的话可以做定量重复性的判定么好友回复:可以的
2010年版药典新增加了很多药物中有机溶剂残留项目,我公司现在仅有一台国产气象色谱仪,没有顶空进样器,据说有手动的仅需几千元,自动顶空进样器国产的要4万,进口的要十几万,不知各位同行现在用的是哪一种?如果用手动的,分析结果能达到要求吗?国产的自动顶空进样器哪种好?欢迎大家讨论!!!
最近我门的DL53电位滴定仪开机后屏幕不停闪烁,没有任何英文字母,一片空白,关机后还闪烁1-2秒,有那位大虾知道是怎么回事?
本人最近在做毕业论文实验(大四) 课题是顶空法测定葡萄酒中的甲醇由于条件限制,只能手动进样,就是水浴箱加热,进样针手动进样仪器岛津GC-2010 顶空条件 70度 恒温 30分钟色谱条件 进样口200度 分流比10:1 柱流量 1.50ml/min柱温 45度4MIN 后5度/MIN到70度FID检测器 温度:220度色谱柱是SUPELCOWAX-10 (30M*0.32mm*0.25um)由于条件限制 只有100ML刻度的顶空瓶 加样量50ML 进样量 1ML问题是:1.对照样中内标(正丁醇)和甲醇的浓度相等(约300mg/L ),可是出峰后甲醇的峰面积很小,大概只有内标面积的0.2~0.4倍,为什么? 2.我用的是震荡水浴锅,但由于水浴锅在8楼,气相在5楼,这来回会不会影响平衡?恳求前辈们帮忙,现在只求能使出峰面积规律一点,不要时大时小,能做一个差不多的标准曲线就能完成论文了。。
本人检测乙烯利,在顶空瓶中与碱反应生成乙烯。条件是恒温70度水浴2h,现想问一下各位大侠,可不可以在2h后把瓶子们拿出,冷却至室温,再依次手动用针来抽气用FID检测器检测。不知道有没有影响?
各位前辈: 有谁用手动顶空做过水质苯系物的啊? 目前就有DB-FFAP毛细柱,HP-5毛细柱,顶空瓶50ml 条件怎样设置啊
我实验室有瑞利公司的SP-2100[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],都是手动进样。现在我想做顶空进样了,请问有方法改吗???
顶空进样手动的隔夜之后进第一针峰特别大,求教大神
请问手动气密进样针如何清洗,顶空用的,谢谢
有没有[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]用手动注射器(抽顶空)能在90度以上使用??哪位大侠推荐一个?
请问顶空手动进样的进样针头要什么规格的好?在哪里可以买到?

星期五(六月二十四号)在一用户处检定一台DDSJ-308A电导率仪:http://ng1.17img.cn/bbsfiles/images/2016/06/201606260553_598169_1626275_3.jpg其它检定项目都较好进行,但检定温度系数示值误差时,我不会手动设置该款电导率的温度为15聂氏度和35聂氏度(25聂氏度可利用拔去温度传感器获得),请教版友上海仪电科技308型电导仪是不能手动设置温度吗?谢谢!
请问各位专家,P-5L2型激光式粉尘分析仪的连续检测一定需要软件控制吗?请问在档位选择里,我选择手动档,其结果一直累计上去,我选择下一个时间段减去上一个时间段的累计值,我就可以得到上一个时间段的累计粉尘浓度,再除以时间,就可以得到其平均粉尘浓度。但是,我发觉这样测试的浓度不稳定,但不知这样测试会不会对仪器的灵敏度和使用寿命有影响?
顶空进样时一直没有溶剂DMF的峰,其它组分的有。但加热后用进样针手动进样时又有。是为什么啊
在质谱中说的自动调谐和手动调谐有什么区别?通常手动调谐时应该如何设置参数呢?当初培训时听的云山雾里的,现在请大家帮忙,谢谢了。
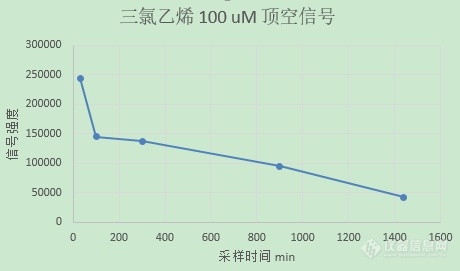
【实验条件】使用40mL聚四氟乙烯隔垫的顶空小瓶,20mL纯水,然后注入50uL的甲醇三氯乙烯溶液注入到水中,使其初始浓度为13 mg/L (三氯乙烯在水中的溶解度100 mg/L左右)【遇到的问题】1. 取样问题使用高鸽阀微量进样器或者普通进样器的时候,需要每次刺破隔垫抽取顶空样品25 μL,然后手动注入到GC中。为了防止刺穿隔垫后堵塞以及用两三次后注射器推杆就会很涩,所以每次取样前我都会使用甲醇清洗微量注射器,但是微量注射器里面的甲醇会影响三氯乙烯的检测浓度,所以【微量注射器中的甲醇】如何除掉呢?2. 三氯乙烯平衡问题三氯乙烯应该迅速平衡(文献上都这么说的),我使用铝盖卷边、丁腈胶塞的棕色厌氧瓶都试过,内部的三氯乙烯顶空气体在24小时后GC峰依然还会降低(我的没平衡?但是不应该漏气才对),并且连续好几次测样误差能在50%-200%,(使用标准气体 甲烷乙烷乙烯手动注入检测误差在5%-10%),这可能是什么原因导致的呢?感谢各位大佬!![img=,460,271]https://ng1.17img.cn/bbsfiles/images/2024/02/202402270950142921_7305_6380806_3.png!w460x271.jpg[/img]
大家平时做样品,每天的样品量肯定都不是固定的,有时多,有时少,样品少就直接做完了手动加载待机程序,让仪器待机,但是样品多时就有可能出现下班时样品还没走完的现象,那你们是会等样走完了,来手动待机,还是直接在批处理后面加上待机程序呢?或许还有其他的方法。http://ng1.17img.cn/bbsfiles/images/2014/10/201410301615_520923_2732869_3.png 我有时不想去实验室时,就直接加载,但是这样也不好,因为晚上扎了针的样品瓶或多或少会挥发些有害物质在实验室,要是再来的话会把样品瓶处理掉,避免这种情况。
手动进样有一定的人为影响因素在内,比如我们在做苯甲酸等样品,也许有的样品只检测出来一点点,我们该如何避免污染呢?哪些操作会带来手动进样器的污染呢?我们又该如何避免?
请问:平面光栅摄谱仪中闪耀角、闪耀波长和仪器的性能有什么联系?闪耀角、闪耀波长的数值是越小越好吗?每台平面光栅摄谱仪中的闪耀角、闪耀波长是固定的吗?
做标准样品测试的时候,FID检测器没有检测到样品峰,不知道是不是进样有问题,所以想试试手动进样,怎么设置? 我把方法里的进样器删除了,运行系列的时候,仪器状态一直是Ready (Standby),不开始程序升温,怎么回事?







 我要推广仪器
我要推广仪器
 下载APP
下载APP