对于高碳不锈钢湿法消解,我们方法是 :盐酸消解1小时后,加3-5ml硝酸,继续加热消解 30min。这样测的 铬含量(标准样)比较理想,但是 其他元素(比如:Si、Ni等)有时候会不平行。求一种高碳不锈钢湿法消解方法:① 保证 Cr 含量测定准确 ; ② 保证其他元素测试结果平行性和准确性。

最近拿到一个样品,高纯石墨,做电池碳芯的那种,不是活性炭。拿来以后试了各种办法,最后终于消解干净,给大家分享一下。第一个图,使用的仪器。http://ng1.17img.cn/bbsfiles/images/2017/04/201704261059_01_3037308_3.jpg第二图,消解时的温度压力显示,最大红外温度240℃,据说,内部温度会更高点,有260℃。http://ng1.17img.cn/bbsfiles/images/2017/04/201704261100_01_3037308_3.jpg这个是用硫酸+硝酸消解出来的,黑乎乎的,跟没有消解一样,其他的酸配比,硝酸,双氧水,什么的基本上都一样。http://ng1.17img.cn/bbsfiles/images/2017/04/201704261102_01_3037308_3.jpg这个使用硫酸+硝酸+高氯酸消解出来的,已经完全消解干净了,没有一点颜色。http://ng1.17img.cn/bbsfiles/images/2017/04/201704261106_01_3037308_3.jpg
最近做了几个高纯氧化铝的样品,摸索了一下前处理方法,跟大家 分享一下。采用铂金坩埚碱融是理想的消解方法,称取1.5g左右的无水碳酸钠,加入约0.2g样品,再加入0.8g硼酸,在1000℃的马弗炉中熔融半小时 以上,45分钟为佳,取出冷却后,用盐酸提取即可。

最近做了一批土壤,先是用的微波消解,原子荧光测汞、砷。结果汞的结果还好,砷的根本不敢看,结果呈两极分化趋势,偏差高达百万倍之巨。实验失败!!!复测的时候,老司机改用水浴方式消解,仍然用原子荧光测,样品结果就具有可比性了,两端极值相差也就是个加倍的关系。即便复测结果也小有问题:与上一年度的结果相比又呈整体偏低的现象。详情请移步到[url=https://bbs.instrument.com.cn/topic/6928587]AFS荧光值严重漂移的可能原因有哪些?[/url]了解,谢谢。另一个值得注意的现象是,经过上次实验后,我们的消解仪似乎被严重污染了——这两天做食品中的铅和镉,发现只要是经过消解的样,铅的样品空白值就特别高;不消解则没有问题。铅空白高,实验猿首先怀疑是新进的电子级的硝酸有问题,但又立即通过对照实验排除。附图数据由PE公司的PinAAcle 900H[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱仪[/color][/url]测试。目前正在处理消解仪,包括消解管、消解罐……正在赶时间做能力验证实验,查不出最终原因很着急的。[img=,632,3451]https://ng1.17img.cn/bbsfiles/images/2018/08/201808191800104944_5969_1838957_3.jpg!w632x3451.jpg[/img]
微波消解仪作为普遍的消解设备,现在的售后服务的效率高不高?存在哪些方面的问题?时间、成本、解决问题的能力等。。
最近做了一些土壤,要求测里面的铜铅锌镉镍铬6种元素;采用的是微波消解(硝酸6ml,盐酸2ml,氢氟酸2ml)--电热板赶酸,ICPMS法测定的方法,标准土样里其余5种金属测定值均能做到质控范围内,但是镍结果高的离谱,一般要比给定值高70%(几次测定平行性较好),不知道这是什么原因,请做过的前辈指点迷津。
测定汞,消解之后赶酸温度到底可以高到多少不会有影响?大家在消解样品赶酸时,温度用的各不相同,不知大家有没有做过相关的试验,验证一下。到底汞可以承受的赶酸温度是多少?赶酸的温度低,自然会是赶酸时间延长,赶酸温度高,可以缩短时间,但是温度过高会有损失,影响测定结果。你是怎么看待这个温度的选择的??你是为了保险起见,低温,延长时间呢?还是高温缩短时间呢?说说你的想法和看法吧!!
目前实验室测定高氯COD用的风冷的消解仪(爱尔的),想换成水冷的,这样冷凝回流效果会好一些,大家有用过爱尔水冷的高氯COD的消解仪没有?好不好用,容易爆管吗?谢谢!
测定砷,消解之后赶酸温度到底可以高到多少不会有影响?大家在消解样品赶酸时,温度用的各不相同,不知大家有没有做过相关的试验,验证一下。到底砷可以承受的赶酸温度是多少?赶酸的温度低,自然会是赶酸时间延长,赶酸温度高,可以缩短时间,但是温度过高会有损失,影响测定结果。你是怎么看待这个温度的选择的??你是为了保险起见,低温,延长时间呢?还是高温缩短时间呢?说说你的想法和看法吧!!
高氯废水COD消解器和COD自动消解回流仪啥区别。买一个行不行。一个能满足实验室需要吗?地表水,地下水,污水前处理都需要啥设备啊?
使用全自动消解仪消解污水:取污水样品50 mL,加入0.60 mL 50 mg/L标准样品,用135℃消解4小时,还剩下5~10 mL的样子,冷却定容至50 mL,样品测定结果为0.010 mg/L,加标样品测定结果为 0.762 mg/L,回收率超过 了120%,请问这可能是什么原因呢?回收率怎么这么高。已经多次出现在此条件下消解出现回收率高的线性,也出现回收率在100±10%这种正常的现象
CEM微波消解仪Mars2,同一批样品6个样2个空白,消解时经常有一个样品温度偏高导致仪器报错自动降温,加的都是5ml硝酸,称样量50mg,不知道哪里出了问题?
[b][[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]搞不定]化工样,消解测定Fe,Ni就是做不好![/b]用强浓酸消解化工产品,最终是钠盐,测定其中Fe,Ni,结果不好:(1)平行很差。特别是Fe,均匀样,仪器,灯都很稳定,有20%波动;(2)比别人低(方法不知道). 做过质控样,标样没问题我只有火焰AAS,没有ICP请问: (1) Na有干扰吗?(2)分光光度是否能做高盐的Fe,Ni? 有干扰吗?谢谢!
按照国标做,曲线做到2.5ng/mL,最高点4600荧光值,空白消解了能有6000多,后来消解过很多次,换了两个厂家的硝酸,基本上都是消解罐直接加酸走消解程序,完了冷却后定容到25mL,空白最低的时候600多荧光值,高的时候8000多。消解罐的处理:第一种 10%硝酸泡,6000、8000、4000啥子值都有,都是特别高 第二种 20%硝酸泡,2000左右 第三种 消解过的罐子清洗干净再加硝酸走消解程序,依然2000左右最低600-800的是一批全新没经过任何处理的罐子,测完一次后想着只加了酸也算是清洗了,就用超纯水清洗了加硝酸微波,完了后反而变成2000了完全不晓得问题出在哪里硝酸直接配置30%浓度的测定正常值,载流本来就是10%的硝酸罐子用8mL硝酸泡了1个小时,不消解,倒出来定容到25mL,仍然不高。想不通为什么非要走了消解程序才会高?有没有大神知道这是怎么回事啊
我无机玻璃用HF消解后在ICP上测试(稀释到100ML),发现外围火焰呈红色,火焰长,接近炬管,害怕烧了炬管。 而波长扫描结果后发现1 PB220背景很高,峰型不太规则(峰高两翼有波浪型干扰),测试结果1PPM的Pb. 2 CD226.502和CD228.802没峰型,但在CD214.438有较规则峰型,在226.510,226.810处有很规则的峰型,背景也很低,不知道样品到底有没有CD呢? 我想知道:无机玻璃是否是高盐样品(我的无机玻璃是无铅的,是什么高钡料玻璃),对这样的样品应该如何测试(稀释更大倍数?)如果是高盐样品,高盐基体对铅等测试的影响有多大?会不会偏差太大?另外,我想知道226.510,226.810是什么元素的干扰峰?高手指教
淀粉检测铅含量,在前处理时,湿法消解空白值高于样品值,排除样品中含量低的原因,还会有哪些原因造成空白高的原因呢??、
我们做土壤中汞,样品空白一直偏高,荧光强度达到1000甚至2000.。。。。质控样品一直做不进去。还有就是消解的三个样品不平行并且同一个样品测定出的样品数据不稳定,变化大。。。大家帮我分析下,先谢谢了
最近做食品中汞的考核做得焦头烂额的,以前从来没做过食品中的汞,一般都是做水。考核样是绿色的,估计是某种蔬菜。我实验室里有一个裙带菜的标准样品,汞的浓度是0.017mg/kg。我前处理的方法是微波消解法,微波之前需要前处理,加入8ml硝酸,先放置过夜,然后在电热板上125度消化1.5小时左右,补加3ml硝酸,1ml过氧化氢,进仪器,微波消解,微波最高的压力是2.0,维持7min。消解完后,取出,电热板125度,赶酸1h左右。测定时用的2%硝酸做载流,0.5KOH+0.05%硼氢化钾做还原剂,不点火,用的冷汞测定。标系以及样品都用5%的硝酸定容,标系做出来是没问题的,线性很好(我用的仪器自动稀释),一般都是3个9,载流空白和标准空白在700左右。但是,盲样的测定结果很郁闷,有以下几个问题:1.样品的空白有时很高有时很低,低的时候减掉载流空白只有200多,高的时候4000多。我使用的玻璃仪器,以及消化管都是用硝酸泡过洗净的。2.已知浓度的样品从来就没作准过,偏高很多倍,5倍以上。不管样品空白是高是低,已知浓度的样品一直偏高。3.样品很多时候都消化不全,前处理到酸都快耗完了,还是有棕色的烟,微波后又赶酸,完了以后消化液里还是有细小的渣,像尘土一样的渣渣。恳请有经验的大侠帮帮忙,帮我分析一下我是哪里没做对,我前后做了快10次了,一次都没做好过,太绝望了,我都快崩溃了,帮帮忙,谢谢!!
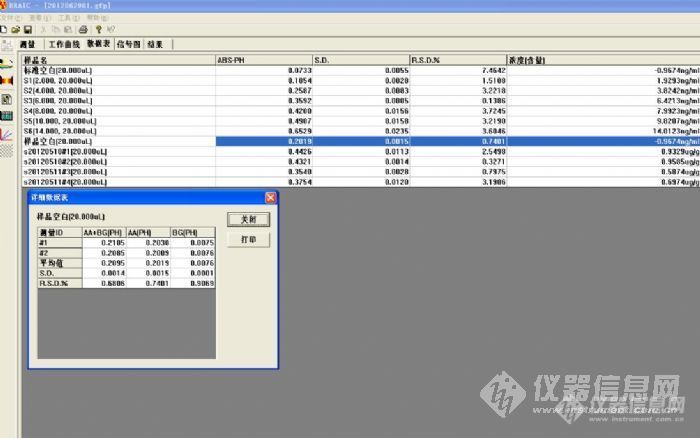
依然是做胶囊Cr 含量,现在仪器稳定了,测量无问题了,开始发现消解罐有问题了:下面是我正常的时候的一张图:http://ng1.17img.cn/bbsfiles/images/2012/07/201207121455_377220_2459406_3.jpg 水正常烧的Abs最高0.01,1%硝酸Abs最高0.07, 当空白样加同等量HNO3消解定容后,空白值在0.20左右,这样看上图得到的结果在4ng/mL左右,结果是0.80ug/g左右。 后面的问题就出现了,有一次消解的时候空白值达到了0.5-0.6,仔细测定了水,1%硝酸的Abs,还有转移定容时候容器的影响,都排除了,就想可能是因为消解罐污染了,因为刚开始用的几次消解空白都很正常,空白罐Abs 0.2,试样罐都在0.4-0.5之间。出问题后的空白都在0.5以上,试样在0.5-0.7之间,结果也受到影响了. 今天想看看消解罐 泡酸后的空白吸收,于是在罐子里放上硝酸,另一个罐子里放水各7mL进行消解. 压力1.0Mpa持续18分钟....悲剧发生了...等消解完后发现装水的聚四氟乙烯罐子内壁凸起了1cm高,好几个凸凸,这个罐子又报废了,才用了几次. 厂家说一个全的罐子需要2500RMB,要这么贵?是我操作失误不该用水,还是质量问题?另外几个罐子都没出问题........ 还有个问题,当试样的含量比较低时候,比我的线性最低浓度还低时候,如果曲线截距较大时候,偏差会大,该如何让这个曲线过0?
我手头没有微波消解装置,脂肪含量高的食品该如何消解?我用高氯酸+硝酸(1:5),在电炉上加热,溶液即将变成无色透明状时,上面还是有大量的脂肪无法消解掉。随后再加热溶液变黑,并着火炸了起来。 请问该怎么解决这个问题?
微波消解法空白试验比样品结果高?是消解罐污染吗?
前几天在做八宝粥钠检测用微波消解样品的时候,准备消解结束的时候突然冒出了一股狼烟,搞得整个仪器室都有一个味道,这是怎么回事呢?消解的酸是硝酸加过氧化氢。请教各位大侠:测钠可以采用微波消解的方法来消解方法吗?有没有什么其他比较好一点的消解方法?
用水监测分析方法第四版的快速密闭反应消解法,加3ml水样用0.2mol/L的消解液,COD测定范围是50-100。测100mg/L标样时,结果偏高,问问各位同行,我是第一次用这个方法测定,请教一下各位同行测定结果偏高哪个这是哪个环节应该注意,然后该怎样调整提高测定精度呢?请多多指教各位同行不胜感激
高铝矾土和氧化铍如何微波消解???
请问一下大虾们。。饰品中的锆石该怎么样消解呀!!!!http://simg.instrument.com.cn/bbs/images/brow/em09511.gif
Mars 5 40个罐的消解样品,加的是硝酸和过氧化氢,消解过后,Pb的空白值和样品值都特别高,平时特别稳定的样品,这次测,都超,平时几十个ppb的,这次测都要1到2个ppm,空白也特别高。硝酸,过氧化氢都用的新的,罐和瓶子都是特别处理的,ms也用百分之5的硝酸洗了一上午,测铅还是很高,是什么原因?有没有什么解决办法
食品行业测汞问题:回收率偏高(140%左右)现有仪器及试剂:优级纯试剂(沪试),科创海光AFS-3100,高压消解罐试验容器均硝酸浸泡、清洗。仪器显著缺点,1.工作曲线拟合只有一次拟合和二次拟合,平且不强制过原点。2.高压消解罐和盖子已混乱,可能密封性不好。详细消化过程按国标GB/T 5009.17-2003(圆大白菜粉,汞含量5-50ug/kg):称量0.3g左右,2个平行样不加标,1个加标10ug/L,1ml 2个样品空白。共5罐,加5ml硝酸(硝酸是不是多了?),加盖过夜,第二天加7ml双氧水。马上放消解罐钢外套拧紧,烘箱(排风系统好像不好了)120度2.5小时。冷却。查看消化好的样品,无色透明!马上用1+9硝酸定容(是不是要赶酸?赶氮氧化合物呢?氮氧化合物怎么赶?引起的干扰是正干扰还是负干扰?),准备测试!达人帮忙看下有何问题啊?
你好,我想问一下,盐酸羟胺溶液是消解前加还是消解后加?盐酸羟胺溶液是不是每次用的时候都需要现用现配?做曲线的时候需要加吗?谢谢
用微波消解法做质控样GSS20共测定结果偏高很多,而且样品空白页很高,荧光值能达到0.5ppb,载流和标准曲线和样品都是用的同一瓶酸,刚打开的,这是为什么呢?求解!!
最近在做个高硅的铝合金,用微波消解后,产品混浊的黑色悬浮物,问怎么才能完全溶解样品







 我要推广仪器
我要推广仪器
 下载APP
下载APP