[color=#444444]我的色谱是GC2020,老化后走空样程序升温时有很多绿色的小杠杠峰,到达终温恒温时有没有小杂质峰,走过多次空样都是这么回事。后来就把衬管和色谱柱都换了,结果还是这样,在程序升温时有杂质峰,到达恒温时没有杂质峰。请高手指教这是怎么回事[/color]
各位老师,我现在在做硝苯地平的有关物质,有一个硝苯地平杂质1的含量忽大忽小,后来我们发现不同的色谱柱,检测出来的含量不同,用了安捷伦、迪马家的C18柱,杂质1含量约0.3%,但是用Waters家的色谱柱,杂质1就是未检出。所以,我想问一下,C18色谱柱中,会不会存在微量物质,促使硝苯地平降解为杂质1。硝苯地平在紫外光破坏、酸破坏、热破坏条件下产生杂质1。另外,单独进杂质1对照品溶液峰面积无变化,每个色谱柱都很稳定。
求啶虫脒水分散粒剂杂质色谱分析的图和分析信息
国内外主要色谱杂志色谱(Chinese Journal Chromatography),中文杂志,双月刊, 1984年创刊Journal of Chromatography (色谱杂志), 国际杂志,30多卷/年,1-3 期/卷,主要以英文发表,1958年创刊Journal of Chromatographic Science (色谱科学杂志),国际杂志, 1963年创刊Chromatographia (色谱法), 国际杂志,以英、法、德文发表, 1968年创刊Journal of Liquid Chromatography (液体色谱杂志), 国际杂志,主要发表高效液相色谱研究报告, 1978年创刊Journal of High Rwsolution Chromatography and Chromatography Communications( 高分辨色谱和色谱通讯), 1978年创刊主要色谱文摘杂志: Gas and Liquid Chromatography Abstracts Gas Chromatography –Mass Spectrometry Abstracts Chemical Abstracts Analytical Abstracts色谱学权威参考书色谱学导论,达世禄编著,武汉大学出版社,1999现代液相色谱,朱彭龄等编著,兰州大学出版社,1994高效液相色谱法,邹汉法,张玉奎,卢佩章编著,科学出版社,2001实用高效液相色谱法,(英)C.F.辛普森,许征帆译,中国建筑工业出版社,1979痕量物质分析[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法,梁汉昌编著,中国石化出版社,2000汪正范等编著,色谱联用技术,化学工业出版社,2001常用中草药高效液相色谱分析,王慕邹等编,科学出版社,1999药物分析方法与应用,马广慈主编,科学出版社,2001室内环境质量及检测标准汇编,中国标准出版社,2003国际上知名研讨会International Symposiumon Column Liquid Chromatography, 1973年开始,两年一次,现在每年一次International Symposia of Chromatography,1962年开始,两 年一次,由Journal of Chromatography杂志出版论文集
药典关于色谱专属性杂质D的表达实在是看不懂:我用的是[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相[/color][/url]1杂质:是对照物自带还是对照物分解的 2杂质含量计算:不知道杂质是什么,怎么计算杂质含量。用对照物和标样的积分对比?3:专属性杂质D在结论中怎么描述?
DB-624 色谱柱分离氯甲烷的杂质: 各位大师,请教个问题,我现在用安捷伦7890配DB-624 分析氯甲烷及其杂质(DME,一氯甲烷,二氯甲烷,氯乙烯,二氯甲烷,二氯乙烯等)的色谱峰及出峰顺序,保留时间最好有啊?现在标准物质没弄着!
有谁能告诉我,用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测农药残留杂质如何去除?我做农药残留用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法,杂质峰很多,标准添加做不出来,谁能帮助解决!!!!谢谢
最近天天发贴求助,就是色谱进样后有时(大部分时间)不出峰,有时候发现灭火,有时候发现进样针堵了,(不知道这些是否是真正原因,这些问题解决后),这些问题解决后接下来就出现长时间大量杂质峰,在检测条件柱温200度,进样200度,辅助箱200度,检测器200度得走三个多小时,其中杂质峰密密麻麻,下一针后还是这样,我的样品是自己配的不可能那么多杂质,这是什么原因呢,期待中~~~
如何用制备色谱柱制备低含量的杂质?
求教气体杂质色谱分析方法
如何用制备色谱柱制备低含量的杂质?
如题。做液相色谱质谱杂质峰丰度很高,达到2*E5,怎么冲都冲不掉,做的是蛋白酶解实验。各位有办法吗?又遇到类似情况吗?
现有一个产品检测项目,按照药典要求,各杂质计算需要采用杂质溶液自我稀释的方法来计算,由于按照药典全套检测,需要大量人力物力。 想请教一下:从GMP角度讲,是否可以直接采集杂质溶液,通过读取色谱图上各杂质的结果来判定盖批次样品是否可以用于小批次混合
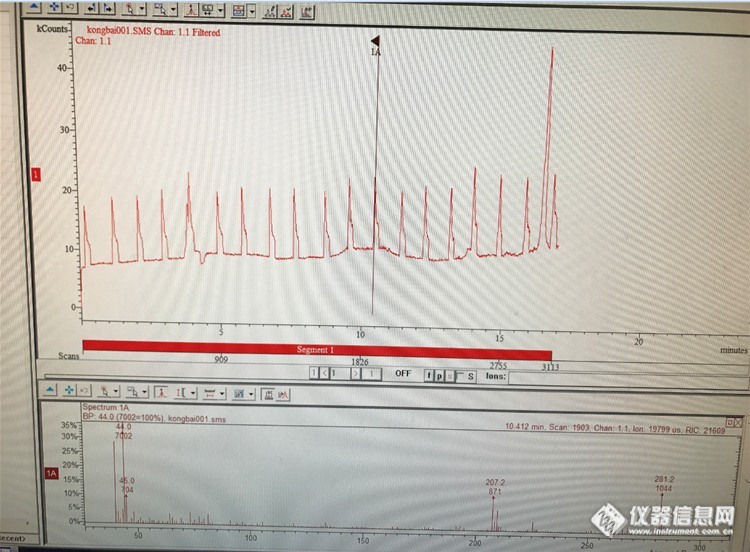
各位老师,我用的是DB624色谱柱,走空白的时候出现这种规律的杂质峰,质谱信息为40,44,207,281m/z。已经排除了隔垫衬管等的污染(更换新的),还是会出现这种情况。是否是色谱柱失效的表现?[img=,690,507]http://ng1.17img.cn/bbsfiles/images/2017/06/201706061738_01_2525115_3.png[/img]
GC分析苯胺及其中杂质的色谱最佳条件是什么?最近杂质环己胺结果重复性特别差!其他组分还行!有高手指点一下吗?
有没有做过维生素A杂质的液相色谱分析?维生素A及其杂质维生素A环氧化物、维生素A醛、维生素A酸没有杂质对照品,以上物质在甲醇-水(90:10)流动相,C18*250mm柱能不能分的开?
[color=#444444]我的一个实验原材料因为是聚合物,购买的原材料浓度应该在95%以上。现在我需要对实验对象中这种原材料的含量进行测定,但打了色谱后,发现原材料那里有杂质峰,推测这个杂质很可能就是聚合物的单体,是不是存在这个杂质峰我就无法做定量分析?请问有什么好办法可以解决这个问题吗?谢谢![/color]
我用的是日立D-2000高效液相色谱仪器 新的 新的waters 氨基柱子 用错了流动相是 乙二胺四乙酸而那钙盐 ,用的时间很短,后来用水冲洗了很久,又用了乙腈。每次出现的是杂质峰,一大一小。太强了信号(700左右),主峰都看不到啊,杂质峰出峰时间固定,面积固定。。现在怎么办啊。。进什么标样出来的杂质峰都一样啊。。一样看不到主峰在哪儿。进样针进行了多次的清洗。进了多次的乙腈都是倒峰。
谁有 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定氯气氢气中杂质含量 的分析方法的相关资料,我从网上搜了一下,都是万方收费的 大家有没有免费的资料 谢谢
各位大侠,请问那个有关于离子膜烧碱项目中用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测氯气和氢气中的杂质气体的方法,介绍一下,定有重谢
求助!!色谱纯丙酮中一般含有那些杂质?哪位大哥能给个色谱图??谢谢了 !!![em0808][em0808]
低分子量肝素钙中杂质的测定用什么色谱柱好[img]http://simg.instrument.com.cn/bbs/images/brow/em09511.gif[/img]
我们用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]面积归一化法测定胺类物质含量,现发现杂质含量偏低(原来正常后突然变低,在其它仪器上测定结果正常),载气压力变大时杂质含量有所增大,不知是何原因,请高手指教!谢谢!
请问气相色谱对气体中杂质的检测能否达到ppb级别?
随着公众对药物安全性的日益关注,控制药物中杂质已成为控制药品质量的关键因素之一,也是困扰着广大药物分析工作者的难题之一。由于药物杂质的来源广泛,已知的杂质可以通过现有的分析手段进行定性定量,未知的杂质则成为分析的难题,因此对于药品的杂质控制首要解决的问题就是将所有杂质进行完全分离。为了让广大药物分析工作者能实现有效地药品杂质控制,全国医药技术市场协会于2012年4月10日-13日在上海市举办“2012药物研究分析中新技术、新方法应用及杂质控制研讨会”。 制药企业和新药研究机构的研发人员,各级药品检验所(院)和口岸药品检验所人员,药品生产企业研发技术与质量管理负责人,新药研发CRO实验室人员及高管,各高等院校、科研院所等相关专业人员100多人参加了此次会议。 在此次会议上,多位行业知名专家钟大放(中科院药物研究所),王洪允(协和医院临床药理中心),胡昌勤(中国食品药品检定研究院),周立春(北京市药检所),王玉(江苏省检品检验所),张尊建(中国药科大学分析测试中心)分别讲解了当前药物分析领域中各种新技术、新方法,探讨分析新技术在药品研发及药品质量控制中的应用,特别是用于生物标志物、活性成份、药物代谢等高通量、定性、定量的各种分析技术,以及新版药典对药物分析方法新要求与国外药典比较等内容。 作为全球色谱消耗品领先的制造商,迪马科技一直致力于为食品、药品检测行业提供完善的技术服务,除与参会专家进行技术交流外,迪马科技技术应用工程师还与广大与会者共同分享了《Dikma 高效液相色谱柱技术应用于药品杂质控制分析》技术报告。 对于药品中杂质控制分析,首先要借助色谱柱进行良好的分离,迪马科技在此次技术报告中重点讲解了在杂质控制中色谱柱的分离性能所起关键作用及迪马科技多款液相色谱柱:Ø Diamonsil(钻石)—通用型反相色谱柱,超高的分离性能特别适合分析复杂的样品及杂质;Ø Spursil(思博尔)—通用型极性改性反相色谱柱,耐受100%水相-100%有机相,特别适用于强碱性化合物和极性化合物的分析;Ø Endeavorsil(奋进)—1.8 μm UHPLC专用色谱柱,超高的柱效满足您UHPLC分离杂质的需求;Ø Leapsil(飞跃)—2.7μm兼容UHPLC/HPLC色谱柱,低柱压设计,高选择性可在HPLC上拥有UHPLC色谱柱的分离能力;[font=Wing
标准品是异黄酮的4种组分,用的流动相是甲醇:水:醋酸=45:54:1,第一和第二种组分的峰型很好,但是第三和第四种组分的峰总是与杂质峰分不开。我也调整了流动相的比例,进样量也调整过了,但是问题依然存在。(我用的液相色谱仪比较老,只能进行等梯度洗脱)。我是新手,对液相色谱了解的也不是很多,现在很着急啊,连标准曲线都做不好。更别说将来要测量样品中的含量了。
请教:液相色谱测定原料药杂质如果结果分别在5%、2%、1%、0.5%、0.1%以内平行结果的相对误差分别应在多少以内?如果是不同仪器、不同色谱柱、同样方法相对误差又该是多少呢?
本人新接触[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url],最近使用的一根毛细管柱子,总是有系统杂质峰,老化色谱柱,烧柱子都没法消除,只有偶尔的时候会莫名其妙没了,这个柱子15M,请问各位有什么办法把处理这个问题,针,进样口没有问题。这些峰在30M柱子不会出现。柱子在刚装上的情路况下,走空针,杂质峰面积可以达到一万多,之后慢慢减小,但是还是有好几百,老化柱子都没法处理,这个样品分析的方法程序升温很快,最慢的也是60,起始温度100。有没有高手解答下?
前几天福立9790气相色谱在正常进行程序升温测样时,突然出了一个5mv左右的杂质峰,空走时也有,目前已经做了以下工作进行排除:1、更换进样隔垫、衬管、清洗FID检测器,做了两次,该杂质峰没有变小的趋势;2、更换色谱柱(柱子型号是SE-54)仍无变化。我也考虑是否是气路的原因,但是这台仪器和另一台9790共用氮气、氢气和空气,而另一台没有这个杂峰。这个杂峰会随着初始温度的变化在不同的保留时间出峰,观察了一下,在230℃的时候会出峰。请问大家有没有遇到过这种情况,或者指点下还有什么原因,感激不尽。
质谱类的高档杂志有哪些







 我要推广仪器
我要推广仪器
 下载APP
下载APP