可否用分析纯的溶剂代替色谱级的溶剂来作为液相色谱的流动相?
可否用分析纯的溶剂代替色谱级的溶剂来作为液相色谱的流动相?
不溶性糖精的高效液相色谱测定方法?请问用高效液相色谱法测定不溶性糖精的具体测量条件,如流动相、流速等,越详细越好,谢谢!!!!
通常进行高效液相色谱分析是优先考虑的是样品不必进行预处理,就可经溶样来进行分析,因此样品在有机溶剂和水溶液中的相对溶解性是样品最重要的性质。由于样品在有机溶剂中溶解度的大小,初步判断样品是非极性化合物还是极性化合物,进而推断用非极性溶解剂戊烷、己烷、庚烷等,还是极性溶解剂二氯甲烷、氯仿、乙酸乙酯、甲醇、乙腈等来溶解样品,并通过实验判断。若样品溶于非极性溶剂,表明样品为非极性化合物,通常可选用吸附色谱法或正相分配色谱法、正相键合色谱法进行分析。若样品溶于极性溶剂或相混溶的极性溶剂,表明样品为极性化合物,通常可选用反相分配色谱法或更为广泛应用的反相键合相色谱法进行分析。若样品溶于水相,可首先检查水溶液的pH值,若呈中性为非离子型组分,常可用反相(或正相)键合色谱法进行分析。若pH值呈弱酸性,可采用抑制样品电离的方法,在流动相中加入硫酸、磷酸调节pH=2~3,再用反相键合相色谱法进行分析。若pH值呈弱碱性,则可向流动相中加入阳离子型反离子,再用离子对色谱法进行分析。若pH呈强酸性或强碱性,则可用离子色谱法进行分析。对呈强离子型水溶性生物大分子的分析仍是高效液相色谱的特殊难题之一,近年随凝胶过滤色谱和高效亲和色谱的迅速发展,对解决像蛋白质、核酸等生物大分子的分析提供了有效的途径。来源:互联网
正相色谱柱内的水分对柱子的分离效果,特别是保留时间,影响很大!好几位老师都这样告诉我。要想稳定,最好是除去其中的水分。问一下,除正相色谱柱内水分的溶剂是什么?用过异丙醇了,效果不好。

液相色谱中追求一个高柱效,高分离是我们做液相色谱追求的目标,但是往往存在很多问题,比如:柱效,分离,拖尾,峰展宽,峰变形,裂缝等等,有些容易解决,有些尝试很多途径都无法改善,当然可能会是方法本身的原因,但是也存在一些忽略的问题:比如本文中提到的溶剂效应,举二例说明: (1)盐酸小檗碱,方法见三黄片补充方法,左图为调整方法前液相色谱图,峰严重变形,我的做法是调大有机相比例,减小进样量,结果出来右边的图1.jpg (5.5 KB)http://ng1.17img.cn/bbsfiles/images/2010/12/201012181618_268050_2019107_3.jpg2010-6-23 23:353.jpg (5.31 KB)2010-6-23 23:36 (2)阿莫西林,方法见中国药典二部,左两图为失败方法,右图为调整溶剂的图:需要说明的是我们的是多元泵,也就是作为溶剂的流动相为量筒配置,试验过程中调整了上机的流动相(有机相减少),而作为溶剂的未调。我的做法是将溶剂的流动相调比例至比上机流动相低5个比例。结果见右图。 2.jpg (9.26 KB)http://ng1.17img.cn/bbsfiles/images/2010/12/201012181618_268051_2019107_3.jpg2010-6-23 23:35 原因,第一例溶剂为甲醇,流动相为乙腈与离子对混合液,第二例溶剂有机相比例比流动相高,共同点为溶剂强度比流动相强。 解析:液相色谱一般进样量为10微升,柱子中局部流动相量比较小(没具体算),当样品溶液进入柱子,10微升体积会改变局部的流动相环境(强度),从而可能出现两种保留行为,反应在图谱上就是变形,如拖尾,裂缝,肩峰。 希望对做液相色谱的朋友有用。
反相液相色谱仪分析中溶质保留值主要由键合相种类、固定相表面积和流动相组成决定。1键合相种类:溶质保留值通常随链长增长或键合相疏水性增强而增大。2固定相表面积:溶质保留值与固定相表面积成正比。当其它条件相同时,溶质在低表面积色谱柱上的保留值小。3流动相组成:溶质保留值可通过改变流动相组成或溶剂强度来调整,溶剂强度取决于有机溶剂的性质和其在流动相中的浓度。
液相色谱中流动相和样品溶剂可否一样?
因为单位只有一台液相色谱,所以大家尽可能条件一致但我的样品与别人选好的流动相不互溶流动相为甲醇与水的混和物我的样品为有机过氧化物及芳香烃类的混合物样品只能溶于甲醇,但不溶于水,也不溶于两者的混合物色谱柱好像是C8所以想知道,用这样的流动相分析对色谱及分析结果有没有什么影响达人指教!
请教液相色谱能否对含有氯化钠的溶液进行分析??? 氯化钠溶液对交换型的固相萃取柱的回收有影响吗?
作为气相色谱的新手,对溶剂的效应十分困惑,谁能详细给我讲述一下溶剂效应呢?先说说我自己的理解,溶剂气话后进入色谱柱,冷凝 ,如果溶剂的极性和固定相相差大,将不能和固定相很好的结合,这将导致溶剂分布较宽,由于样品分布在溶剂里面样品也会展宽。其次,冷凝是部分冷凝怎么办,会不会一部分液态保留了,气态的跑了。希望高手能为我解惑。
用于液相色谱的各种溶剂的性质[~95083~]
[color=#444444]我在做合成实验中,用不同的溶剂去溶解样品,做高效液相色谱分析,流动相为甲醇和水,发现有的溶剂会出现溶剂峰,有的溶剂就不会出,这是为什么呢?求大神指教迷津,另外有没有相关专著讲述HPLC中溶剂峰出峰时间的[/color]
小白求助:使用液相色谱,样品的溶剂需要与做标线的标液溶液完全相同吗?如果不相同,会影响出峰和积分结果吗?比如说,我的标液是乙腈中的,流动相也是乙腈和水,我现在接到一个甲醇或二氯甲烷等其他溶剂的样品,我需要换标液,换方法,重新做线,还是可以直接进样?
请问下液相色谱可以测试碱性溶液吗?含氢氧化钠0.04%。主要想测试里面其他的有机组分。
[color=#444444]做液相时,完全相同的条件。第一天色谱图好好的,第二天开始色谱峰全集中在溶剂峰处了,且强度很强。这是咋回事[/color][color=#444444]图一是正常的。图二是不正常的[/color][color=#444444][img=,690,510]https://ng1.17img.cn/bbsfiles/images/2019/07/201907111117570447_2503_1848218_3.jpg!w690x510.jpg[/img][img=,690,510]https://ng1.17img.cn/bbsfiles/images/2019/07/201907111117573610_6815_1848218_3.jpg!w690x510.jpg[/img][/color]
请大家谈谈液相色谱中样品溶液的浓度问题,已同事说可以看色谱图中的峰面积,差不多在7000000左右,还有就是响应值在500mV左右,他说的有道理吗?还是必须要做浓度线性关系呢?
各位大侠,用液相色谱做样品时,对样品中的溶剂和我使用的稀释剂有要求么?有不能使用的试剂么,类似于二氯甲烷,四氢呋喃,丙酮等?如果使用那些溶剂对机器有什么影响?
[color=#444444]我想用液相色谱测量牙膏中的EGCG,但是含量较少,所以我想浓缩这个EGCG来测,有没有办法用4L溶剂溶解50g牙膏的方法呢,要充分溶解的,谢谢各位了[/color]
高压液相色谱 样品用什么溶解 问题补充:样品 用乙腈甲醇 不能很好的溶解 怎么办啊
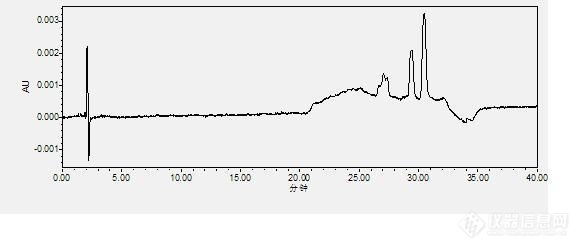
[color=#444444]最近用了一个方法去测产品中有关物质的情况,色谱条件:[/color][color=#444444]醋酸盐溶液(pH=8.0)的配制:0.7708g(0.01mol)醋酸铵,用水溶解稀释至1000ml,用氨水调pH到8.0[/color][color=#444444]流动相A:乙腈:醋酸盐(pH=8.0)/25:75[/color][color=#444444]流动相B:乙腈:醋酸盐(pH=8.0)/60:40[/color][color=#444444]色谱柱:C8(5µ m,4.6×250mm) 流速:1.5ml/min 波长:280nm[/color][color=#444444]梯度:Time(min) A% B%[/color][color=#444444]0 100 0[/color][color=#444444]18 100 0[/color][color=#444444]23 50 50[/color][color=#444444]30 50 50[/color][color=#444444]32 100 0[/color][color=#444444]40 100 0[/color][color=#444444]用流动相B作为溶剂,进了一针,色谱图如下:[/color][color=#444444]在25min到32min之间有干扰峰,按理说,应该没有的。为什么呢?各位大哥大姐又遇到这种情况的吗?[/color][color=#444444][img=,581,250]https://ng1.17img.cn/bbsfiles/images/2019/10/201910101057227722_8944_1806906_3.jpg!w581x250.jpg[/img][/color]
在做液相色谱测定天然提取物测定时用甲醇水系统不能将其峰分开,需要加缓冲溶剂,反相高效液相通常加什么缓冲剂,怎么配制?
有人说液相色谱所用的有机溶剂,如甲醇、乙腈等,要用进口的,这样不会有杂峰,本底也低,这样对吗?国产的哪个牌子最好。
请问大侠们,如何测定香精中未知增溶剂?(定性&定量)如果已经知道是PC309,用液相色谱怎么定量?P.S. 没用过液相色谱,只用过GC和GCMS。。。
我是大学生,最近学习气相色谱。我用的溶剂是乙酸,条件是厂家给的条件:柱箱温度:190度,气化室温度:300,检测室温度:280,厂家用的色谱柱是OV-17毛细管柱。我学校的色谱柱是30QC3/AC20 0.5。我是按照这些条件做的,进样量0.1/0.2uL。出来的色谱图只有溶剂峰能和厂家给的色谱图对上,其他的峰都没有出来,只有许多很小的峰,而厂家给的的谱图中原料的峰也是很大的,可我却怎么也弄不峰来。溶液的浓度我试过了,从低到高,基本都一样,只出溶剂峰,其他就是一些很小的峰。我想请问大家,我应该从哪些方面解决这个问题呢?色谱柱不行?这两个差别大吗?条件不合适?谢谢大家能够给我提供宝贵意见!
[color=#444444]环己胺一般用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析,但是环己胺的水溶液不能进[url=https://insevent.instrument.com.cn/t/Mp]气相[/url],可以进液相色谱吗?会出峰吗?如果萃取环己胺水溶液该怎么萃取?谢谢[/color]

[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=171677]液相色谱溶剂系统的选择与优化1[/url][img]http://ng1.17img.cn/bbsfiles/images/2009/09/200909180921_171680_1608119_3.jpg[/img]文件合并见6楼
[align=center][b]谈谈溶剂对色谱柱的影响[/b][/align][b]毫无疑义,溶剂系统对液相色谱分析有重要的影响。现在有商品化的色谱纯溶剂,极大的方便了日常的检测工作。但是我们经常可以看到这样的描述,在检测开始前,色谱纯溶剂要经过微孔滤膜进行过滤。这样做有意义吗?能够真正起到保护色谱柱的作用吗?对于色谱纯溶剂,有国产和进口之分。国产色谱纯溶剂的质量良莠不齐,用微孔滤膜过滤是必须的。我以前听企业的人说,国产的色谱纯溶剂容易堵色谱柱,就是用微孔滤膜过滤以后效果还是不好。其实大家一直忽略了一个问题,就是在过滤的过程中,多次转移造成的交叉污染是不可控制的。但是对于大品牌进口溶剂来言,我认为通过微孔滤膜过滤不仅不能起到过滤的目的,反而会引进污染,往往是越过滤越脏。通常的溶剂过滤系统都是开放的敞开体系,在溶剂过滤的过程中过滤瓶的瓶壁,空气中的尘埃,溶剂转移过程中造成的交叉污染都有可能将污染物带入溶剂系统中。而我们知道,进口溶剂的蒸发残渣不会超过3ppm,这种影响对于色谱柱本身而言,相比于交叉污染的影响来说,是可以忽略不计的。另外,色谱柱本身也是耗材,自身是有使用寿命的。我们能够做到的就是好马配好鞍,这样才能最有效的提高色谱柱的使用寿命。 溶剂系统除了色谱纯的溶剂之外,还有水,做液相色谱的时候,我们通常需要引入水相作为溶剂系统。有的人会将超纯水通过微孔过滤系统进行过滤处理。这种处理有意义吗?有必要吗?超纯水在经过处理以后,已经具备了直接进液相色谱的条件了,再次处理无异于画蛇添足。反而非常容易将尘埃、微生物引入色谱系统中。笔者认为,最好的超纯水系统是用蒸馏水做水源,进行二次处理。对于溶剂系统,您有什么看法,希望您可以发表不同的意见和看法,共同学习,共同进步![/b]
做液相的,都知道做实验时有机溶液带来的危害。不知道有没有一种简单、快速的方法,判断液相色谱室里的有机溶液溶度较高了,已经能对人体造成一定伤害?

本人使用的是岛津10A系列的液相色谱仪,在进行试验时发现3分钟左右有一个峰经常存在,影响有关物质的测定。我使用的溶剂是流动相,在进溶剂时,该峰的峰面积不随进样体积的变化而变化,其他溶剂峰大小会有变化;在进空针时,没有溶剂峰出现,可以判断排除是系统的干扰,考虑是进样器带入的东西,更换进样垫后,这个峰仍然存在,在此请问各位大虾,关于这个溶剂峰的可能还有哪些来源呢?不同进样体积的溶剂峰如下所示,红圈的为那个鬼凤点击打开链接点击打开链接点击打开链接file:///c:/documents and settings/caoguifang/application data/360se6/User Data/temp/2014111914375559.jpg







 我要推广仪器
我要推广仪器
 下载APP
下载APP