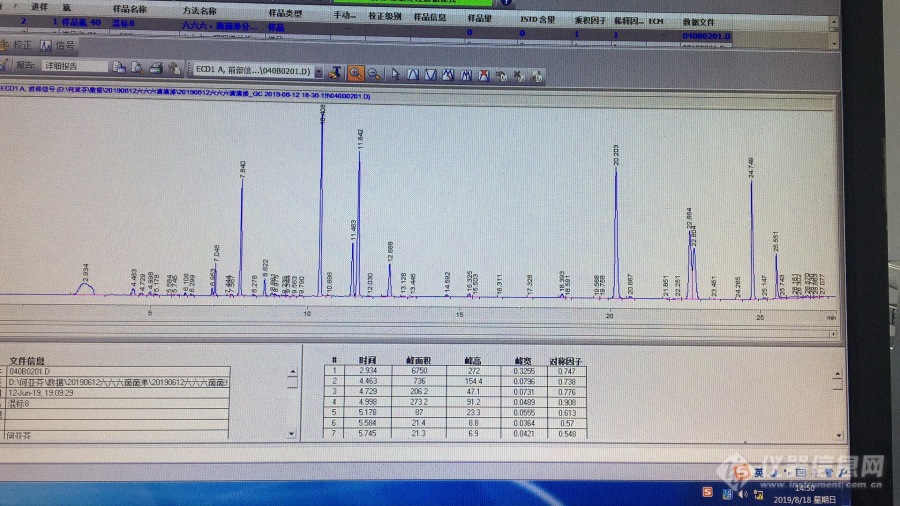
仪器是安捷伦7890A[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url],ECD检测器。检测氯氰菊酯和高效氯氰菊酯、氟氯氰菊酯和高效氟氯氰菊酯、氯氟氰菊酯和高效氟氯氰菊酯。检测方法:NY/T761-2008。仪器条件,柱温200℃,检测器320℃,流速1.0mL/min,柱子HP-5,柱温150℃(2min),6℃/min→270℃(8min)。氮气5个9以上,进样口的衬管与隔垫更换新的,柱子两端也截过。进样1uL,标品浓度200ng/mL。只有溶剂峰,什么峰都没有。但是同样的仪器条件,能做出666、滴滴涕。有没有和我遇到同样的问题的?是怎么解决的呢?图1是之前做666、滴滴涕的图谱图2是这两天做菊酯类农残的图谱[img=,690,387]https://ng1.17img.cn/bbsfiles/images/2019/08/201908181459474438_2497_2450726_3.jpg!w690x387.jpg[/img][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/08/201908181500312972_3742_2450726_3.jpg!w690x517.jpg[/img]
在ECD上走三氟氯氰菊酯标液出现三个峰,保留时间分别是16.05、16.14、16.26,检测样品时在三氟氯氰菊酯出峰的地方出现两个峰,时间分别是16.05、16.26。是否可认为该样品中含有三氟氯氰菊酯?
茶叶氯氟氰菊酯和高效氯氟氰菊酯2763中要求按照SN/T1117检测,但是SN/T1117-2008已经废止,并且没有替代标准。请问茶叶氯氟氰菊酯和高效氯氟氰菊酯可以用什么标准检测?是否还可以用2763判定?
请问下我现在用气相检测氟氯氰菊酯,有时出三个峰有时会有第四个峰?样品中好像还有氯氰菊酯,而且两个物质都是三个峰,连的很近,有什么方法可以分开一点吗?
用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]检测果蔬中菊酯类(氯氰菊酯、氟氯氰菊酯、氯氟氰菊酯,氰戊菊酯,溴氰菊酯)农药,刚开始进标准混合溶液时,它们都有出峰的,可是再进了十几个果蔬样品后,再重新进标准溶液后,质控的标准溶液就不出峰了,也就是说后面出峰的菊酯类走着走着就没有响应了,前面出现出峰的比如有机磷等出峰还是正常的,然后重新老化了VF-1701MS色谱柱,后面出峰的菊酯类标准溶液还是没有出峰?现在要如何解决?

我要检测硫丹(包括:1-硫丹,2-硫丹,硫丹硫酸盐),甲氰菊酯,氯菊酯,氟氯氰菊酯,这6种物质,由于我不清楚这6种物质在我的气相上的大致出峰时间,所以我决定用全扫描的方式试下,程序升温的方法:40度保持1min;然后以30度/min升至130度,再以5度/min升至250度,最后以10度/min升至300度,并保持5min,后运行5min,以去除残留于色谱柱中的杂质。色谱柱:HP-5ms;溶剂延迟时间:5min,从5min开始至35min,SCAN从50到500,我的TIC图是这样的:http://ng1.17img.cn/bbsfiles/images/2016/07/201607140743_600414_2166779_3.jpg局部放大:http://ng1.17img.cn/bbsfiles/images/2016/07/201607140743_600415_2166779_3.jpg对着一个峰右键双击,看其碎片:http://ng1.17img.cn/bbsfiles/images/2016/07/201607140744_600416_2166779_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/07/201607140744_600417_2166779_3.jpg
对于保鲜生姜和大蒜用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]检测高效氯氰菊酯和三氟氯氰菊酯,样品应怎样处理?用ECD检测的话。
请问氰戊菊酯、溴氰菊酯、三氟氯氰菊酯用液质检测应该选择哪些离子对?我的液质型号是安捷伦液相1200-AB质谱3200Q trap,选择上述菊酯的分子量为母离子,扫Q1的时候响应不高,打碎后MS2也找不到子离子,跪求高手指教!
[em09512]请问有没有人做过生姜中三氟氯氰菊酯的检测?有谁做过请指教一下,万分感谢!
请问下,我现在用气相检测氟氯氰菊酯,有时出三个峰有时会有第四个峰?
氯氟氰菊酯用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用仪[/color][/url]检测,连进三针数据越来越大,但是同一个小瓶里面的毒死蜱却很稳定。
氟氯氰菊酯用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]检测,母离子与子离子是多少?用的是AB4500,DP与CE各是多少?
想请问各位大侠:5.7%的氟氯氰菊酯农药检测方法?用液相色谱可以做吗?具体检测条件是怎样?流动相用正己烷和乙腈吗?
今天领导忽然问起了专业性话题,说我们平时检测的氯氰菊酯有包括高效氯氰菊酯和顺、反氯氰菊酯吗?一下子我蒙了,我只知道是氯氰菊酯还真不知道有没有包括以上两种。就此问题也咨询一下大家了,我还真搞不太清楚,高效氯氰菊酯和顺、反氯氰菊酯有单独的标准品吗,与氯氰菊酯是不一样的东西吗?望赐教,小女子实感惭愧,但得不耻下问啊!
大家好,用HP-5MS的色谱柱、GCMS来检测氯氰菊酯与氟氯氰菊酯,由于氯氰菊酯-4与氟氰戊菊酯-1无法分开,但是它们的检测的碎片不一样,氯氰菊酯的碎片为163、165、181;而氟氰戊菊酯的碎片为199、157、451,是否一定要将这两种物质分成两组分别进行检测,有人将它们两个物质放在同一组中进行检测的吗?如果放在一起检测,在定量上会有干扰吗?

请问下我最近用气相做氟氯氰菊酯,可是有时有出来四个峰http://ng1.17img.cn/bbsfiles/images/2013/11/201311212048_478686_2773917_3.png积分也是分开的http://ng1.17img.cn/bbsfiles/images/2013/11/201311212050_478687_2773917_3.png有时候又是三个峰http://ng1.17img.cn/bbsfiles/images/2013/11/201311212051_478688_2773917_3.png不知道是什么情况!请大家帮着分析下!
有谁参加了这次玉米中的溴氰菊酯、氯氰菊酯残留检测的比对,可以交流一下吗?我们做的初步结果为1号样为100、200ppb,2号样为50、100ppb,但是我们的回收率较高,约为130%,有朋友能交流吗?
我用的是Agilent 7890A,ECD检测,HP-5,30m×0.32mm×0.25um的柱子,用了很多程序升温方法都无法分离氯氰菊酯和氟氰菊酯(即氟氰戊菊酯)。[em0702] 请问版上的各位兄弟有没有出现过该问题,即氯氰菊酯四个峰中的最后一个峰与氟氰菊酯的第一个峰重合(经单标进样后确认的),各位是如何通过程序升温或其他方法将此两种农药分开的?能否赐教,不胜感激[em0706]
问题:高效氯氟氰菊酯和氯氟氰菊酯,分子量一样,CAS号不一样,该怎么判定回复:CAS号不一样,说明他们不一样,有可能是取代基位置不同大家怎么看
[b][color=#444444]食品中的氯氰菊酯、氰戊菊酯残留量检测需用何种色谱柱?[/color][color=#444444]现GB/T 14929.4—1994和 SN 0217—93的检测方法所要求的色谱柱类型不一样,不知要采用何种色谱柱比较[/color][/b]
我想问下各位老师,如果测蔬菜中氯氰菊酯的残留,是买氯氰菊酯和高效氯氰菊酯的标准品,貌似氯氰菊酯包括了高效氯氰菊酯,总共有8种异构体,而高效氯氰菊酯有4种异构体,现在市面上的氯氰菊酯标准品和高效氯氰菊酯标准品是否含量相同(比如坛墨1000μg/mL的)?如果含量不同,那么是偏向哪个多一点吗?计算含量需要加和还是将几个峰合在一起积分呢?
哪位做过蔬菜,水果中的高效氯氰菊酯残留检测?能分享一下检测标准吗?万分感谢!!!!
做有机氯,10PPb的α-666进样4微升,信噪比只有100;溴氰菊酯850μg/L,进样2微升,信噪比只有60;东西4000A前开门降温色谱(可能很少人听说过色谱柱箱降温必须打开柱箱门,由于无后门自动控制)。都采用不分流,加脉冲后,基线电压差满足说明要求。计算出的检测限远高于国标上的检测限,柱子是新柱;是什么问题??气化管使用没多久(应该硅烷化处理过???),是设备灵敏度就是这样还是其他???
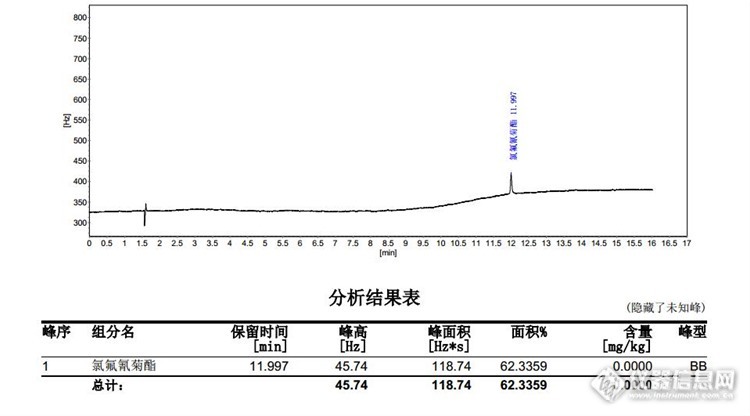
简单点说就是6月份做一个高效氯氟氰菊酯的平行样品,测出来浓度约为0.08ppm然后7月份再重测发现浓度在0.12ppm左右。由于机器和其他原因请不要吐槽为什么0.25mm的-35柱子要用3ml/min的流量,更不用提为什么不分流进样峰面积都这么小。。。。[img]http://simg.instrument.com.cn/bbs/images/default/em09509.gif[/img]两次条件分别如下6月进样口 250 ,不分流进样,色谱柱 EC-35, 柱流量3ml/min程序升温 150以15℃/min至210以10℃/min至260以20℃/min至300保持5min检测器温度 3207月进样口 250,分流5:1,色谱柱DB-1MS,柱流量1ml/min程序升温 150以15℃/min至210保持2min,以8℃/min至260保持2min,以10℃/min至300保持7min检测器温度 300=======================================================================================目前我所能想到的原因如下1.样品瓶虽然有换新盖子然后保存,但是溶剂依然有损耗导致浓度变高,或者其他物质分解形成共流出化合物(我有仔细检查样品瓶溶液高度,感觉不太可能)2.观察色谱图,由于机器和一些其他原因,6月份出峰面积很低,0.2ppm标液峰面积在100左右,样品实测约40峰面积,峰面积太小导致误差变大(我有做平行,平行性感觉勉强达到要求,即使有误差也不会从0.08变到0.12的地步)3.查资料得知高效氯氟氰菊酯在275℃左右分解,6月份出峰时间在12.0左右,此时柱箱温度300,高效氯氟氰菊酯部分分解。相比而言7月份出峰时间在14.3,此时柱箱温度260未达到分解温度4.6月份高效氯氟氰菊酯标液出峰仅一个,且基线杂质较少。7月份高效氯氟氰菊酯出峰两个,且基线杂质较多。考虑相似相溶原理,理论上用-1柱子会有更好的分离和出峰效果,这样也就是说在使用-1柱的时候会有部分基质中的杂质形成共流出化合物影响检测结果。而在-35中这部分杂质溶解效果不好不出峰。5.6月份样品检测中在目标物前面有几个很微小的峰(或许增大灵敏度就能出来),我有考虑过是不是就是高效氯氟氰菊酯本该出几个峰,但是标液中仅有一个峰,或许是我所用标液与样品中的高效氯氟氰菊酯不一致?他的里面含有较多的其他的同分异构成分?目前我比较偏向第四种原因,因为比较色谱图,7月份高效氯氟氰菊酯标液的图谱两个峰比例与样品中比例不一样,也就是说很有可能是其他物质在相同时间出峰影响了检测结果。但是这又带来一个新的问题,按照色谱原理根据相似相溶在做有机氯农药的时候都是推荐非极性的柱子,但是遇到以上这种情况岂不是恰恰用其他极性的柱子会更好的排除干扰?===========================================================================================色谱图缩略如下6月0.2ppm标样A[img=,690,382]http://ng1.17img.cn/bbsfiles/images/2017/07/201707141052_01_2972029_3.jpg[/img]6月样1-A[img=,603,367]http://ng1.17img.cn/bbsfiles/images/2017/07/201707141054_01_2972029_3.jpg[/img]7月0.2ppm混标[img=,690,276]http://ng1.17img.cn/bbsfiles/images/2017/07/201707141054_02_2972029_3.jpg[/img]7月样1[img=,690,445]http://ng1.17img.cn/bbsfiles/images/2017/07/201707141054_03_2972029_3.jpg[/img]
第一组,氯氰菊酯和高效氯氰菊酯,GB2300.8种查到氯氰菊酯的离子为181,152,180但是没找到高效氯氰菊酯,找到了一个顺式-氯氰菊酯离子为163,181,165.这个顺式-氯氰菊酯是高效氯氰菊酯么,但是英文名对不上啊,氯氰菊酯的四个峰是不是包括了高效的,求高手解答。第二组,氟氯氰菊酯和高效氟氯氰菊酯,23200.8只查到了,高效的181,197,141氟氯氰菊酯没有找到,氟氯氰菊酯又名三氟氯氰菊酯,能不能用高效的离子呢?
各位大侠,有没有人知道国标中蔬菜中的氯氰菊酯、毒死蜱、乐果等的检测方法,最好是经过验证改良的,因为如果完全按照国标来做的话,好象效果不是很好。很急哦,谢谢大家献策[em31]
各位大侠,有没有人知道国标中蔬菜中的氯氰菊酯、毒死蜱、乐果等的检测方法,最好是经过验证改良的,因为如果完全按照国标来做的话,好象效果不是很好。很急哦,谢谢大家献策
[b][color=#333333]高效氯氰菊酯,[/color][color=#333333]ECD[/color][color=#333333]检测柱温箱温度至少多少啊?[/color][/b]
个物质的紫外吸收光谱,在不同的液相仪器条件下,是一样的吗?在国标里高效氯氟氰菊酯的检测波长是278nm, 但是我在实际做的过程中,发现210nm吸收值最大, 选择检测波长的依据是什么?不过国标里的流动相条件是:正已烷:四氢呋喃=99.3:0.7我用的条件是:正已烷:异丙醇=90:10,色谱柱不一样。
安捷伦[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]做嘧霉胺,氟虫腈,虫螨腈,甲拌磷砜,啶虫脒,氯氟氰菊酯,吡菌磷,哒螨灵,苯醚甲环唑的时候碰到的若干问题。仪器条件如下进样量:1ul。进样口温度:290℃。进样方式:无分流进样,1.5min后打开分流阀和隔垫吹扫阀。色谱柱型号:HP-5。载气:氦气,纯度≥99.999%,流速1.2ml/min。电子轰击源:70eV。离子源温度:230℃。GC-MS接口温度:280度。选择例子监测:每种化合物分别选择一个定量离子,2个定性离子。色谱柱温度程序:40℃保持1min,然后以30℃/min程序升温至130℃,再以5℃/min升温至250℃,再以10℃/min升温至300℃,保持5min。问题一,氯氟氰菊酯的定性离子。做添加回收的时候,标准曲线的比值和添加样品的比值相差很大,标准的定性离子的比值大约是26和75左右,添加样品就去到160和180左右。想知道哪里出现了问题,或者加入没有操作错误的话,怎么处理。[img=氯氟氰菊酯,690,374]https://ng1.17img.cn/bbsfiles/images/2019/09/201909021112439548_8430_2342324_3.jpg!w690x374.jpg[/img]问题二,甲拌磷砜,跑样品空白的时候,定量离子有检出,但是定性离子比值匹配,我就认为这个峰不是甲拌磷砜,删除了。但是在分析加标样品的时候,基本回收率都超高,我怀疑样品空白的那个定量离子和我的加标峰叠在一起了 这个怎么处理。







 我要推广仪器
我要推广仪器
 下载APP
下载APP