ABI基因分析仪是测什么的?它和[url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]$$PCR[/color][/url]荧光定量有什么区别呢?

如下手持式基因与核酸扩增分析仪的市场需求怎么样?[img=,690,975]https://ng1.17img.cn/bbsfiles/images/2019/07/201907121534081873_2288_2963345_3.jpg!w690x975.jpg[/img]
远志系陕西道地药材,是“秦药”大宗道地药材品种之一[1]。《中国药典》2020年版所收载的远志为远志科(Polygalaceae)植物远志Polygala tenuifolia Willd.或卵叶远志P. sbirica L.的干燥根[2],具有镇静安神、祛痰开窍、解毒消肿等功效[3]。现代研究表明,远志的主要活性成分有皂苷类、寡糖酯类、酮类等,具有抗记忆障碍、保护中枢神经系统、抗抑郁、抗心肌缺血和抗肿瘤等作用[4]。目前,关于远志的研究多集中于含量研究[5]、活性测定[6]、遗传多样性分析等[7]。随着分子生药学的发展,对药用植物相关活性成分生物合成途径相关调控基因、转录因子的挖掘已成为研究热点,基因组学、转录组学等技术在远志上的成功应用,也为远志基因家族的筛选、鉴定与分析提供了技术支撑和数据基础[8]。 碱性亮氨酸拉链(bZIP)基因家族作为真核生物中转录网络的重要开关,是植物中最大的转录因子家族之一。bZIP结构域由两个区域组成,即DNA结合基本区和亮氨酸拉链区[9]。bZIP基因家族成员通过差异基因网络或生物过程,在调节植物发育、生长以及盐胁迫响应等方面发挥着重要作用[10]。研究表明,拟南芥Arabidopsis thaliana L.、番茄Solanum lycopersicum L.、黄瓜Cucumis sativus L.、李子Prunus salicina L.和蓖麻Ricinus communisL.等多种植物中的bZIP参与调控组织分化、细胞生长、糖代谢、生物和非生物胁迫等多个生物学过程[11-12]。bZIP基因家族成员还参与多种药用植物次生代谢产物合成调控,如丹参Salvia miltiorrhiza Bunge.的SmbZIP1基因可抑制丹参酮的积累,大豆Glycine max (Linn.) Merr.的GmbZIP123基因则参与大豆种子脂质积累的调控[13]。同时,bZIP表达受外源激素和胁迫诱导,壳聚糖处理葡萄Vitis vinifera L.12 h下,其VvLysM8和VvLysM9基因表达量显著提高[14],糜子Panicum miliaceum L.中的PmbZIP97不仅受到脱落酸(abscisic acid,ABA)、盐和干旱胁迫强烈诱导且参与调控萌发后的根系生长[15]。 本实验利用远志三代转录组数据,以bZIP基因家族为研究对象,对其基因家族进行成员鉴定和生物信息学分析,并确定其在远志中的结构特点与进化特征,进一步通过实时荧光定量分析其在不同组织、不同处理条件下的表达模式,为后续深入研究bZIP的生物学功能奠定基础,同时为bZIP家族可能参与远志次生代谢成分生物合成途径研究提供思路。 1 材料及仪器1.1 材料2021年10月于陕西中医药大学药用植物园(陕西咸阳)采集3年生远志Polygala tenuifolia Willd.及其成熟种子,经陕西中医药大学杨新杰副教授鉴定。选取5株三年生长势均匀的远志植株,将根、茎、叶等量混合后进行全长转录组测序分析。1.2 试剂及仪器ABA、壳聚糖(chitosan,CHT)均购自上海源叶生物科技有限公司,Trizol总RNA提取试剂盒、dd H2O均购自生工生物工程(上海)股份有限公司,TB Green® Premix ExTaqTM Ⅱ (TliRNaseH Plus)、PrimeScriptTM Ⅱ 1st strand cDNA Synthesis Kit购自TaKaRa公司(日本),所用引物由武汉金开瑞生物公司合成。StepOnePlusTM Real-Time PCR(qPCR)仪(美国Applied Biosystems公司),NanoDropTM 2000分光光度计(美国Thermo-fisher公司),K5800自动检测超微量分光光度计(凯奥公司),?80 ℃超低温冰箱(中科美菱公司)。 2 方法2.1 样品的处理选择大小均一,颗粒饱满的远志种子,用自来水冲洗1 d,10%双氧水消毒,播种于装有泥炭土的花盆中,在光周期16/8 h,光照强度9 000 Lx条件培养[16]。选取长势均一的2月幼苗,喷200 μmol/L ABA、200 μmol/L CTS,干旱(10% PEG 6000)、盐(100 mmol/L NaCl)20 mL,以无菌水作为对照组;以0 h为空白对照,重复3次,6、12、24和48 h取样处理(3株),于?80 ℃冰箱储存,采用PacBio Seque Ⅲ进行上机测序,获得远志全长转录组学文库[17]。2.2 远志bZIP家族基因鉴定及理化性质分析基于远志转录组数据库,筛选出注释结果为bZIP的序列,将序列gene id对应的fasta结果输入editseq软件,进一步获得具有完整开放阅读框(open reading frame,ORF)的基因,通过NCBI中的BlastX进行比对与鉴定。Protparam分析目标蛋白的理化性质,ProtScale预测不同氨基酸中的蛋白亲疏水性[18]。2.3 远志bZIP家族基因二级结构、信号肽、跨膜结构及亚细胞定位分析用ExPASy分析基因编码蛋白质的结构域,CDD验证;ProtParam和SOPMA分析远志bZIP转录因子的二级结构;SignalP-5.0和TMHMM预测信号肽和跨膜区域;WoLF PSORT预测亚细胞定位[19]。2.4 远志bZIP家族基因进化树构建从Tair网站下载拟南芥蛋白序列,通过MEGA软件对远志、拟南芥bZIP氨基酸序列进行多序列比对,利用MEGA的最大自然法构建系统发育树,重复次数设置为1 000次[20]。2.5 远志bZIP家族基因密码子偏好性分析及蛋白互作预测分析采用CodonW、CUSP和Chips分析密码子偏好性。蛋白互作预测分析利用STRING进行,并以拟南芥筛选其同源基因后,通过Cytoscape 3.9.0软件作图。2.6 远志bZIP家族基因蛋白特征、保守基序分析及不同组织表达量热图通过chiplot分析bZIP蛋白的结构域,MEME获得bZIP蛋白的保守氨基酸基序,并用TBtools进行可视化,Weblogo分析蛋白序列位点。利用诺禾云平台将转录组数据库中27个PtbZIP基因在远志根、茎、叶3个部位的差异表达数据进行层级聚类分析。2.7 远志bZIP家族基因表达模式验证与分析Trizol法提取各样品总RNA,凝胶电泳检测后测定总RNA浓度。使用Prime Script TM II 1st strand cDNA Synthesis Kit合成cDNA,检测浓度后于?20 ℃保存备用。设计荧光定量引物,并送生工生物工程(上海)股份有限公司合成。以甘油醛-3-磷酸脱氢酶(F:5’-ACAGCAACGTGCTTCTCACC-3’,R:5’-CCCTTCATCCACCACCGACTA-3’)为内参基因,验证PtbZIP26(F:5’-GCACTGATGG- GAAGGCTGAA-3’,R:5’-GATTGCCCAACAC- TTGAGGG-3’)、PtbZIP27(F:5’-GTCGGATGGT- AGTGAACGGG-3’,R:5’-CACCATTTCCCGAAC- CCTGA-3’)在不同部位样本中的表达量。选择表达量较高的PtbZIP26进行不同激素、胁迫处理下的表达量分析。qRT-PCR反应体系为TB Green Premix Ex Taq Ⅱ(2×)5.0 μL;上下游引物各0.4 μL;50×ROX Reference Dye 0.2 μL,cDNA 1.0 μL;ddH2O 3.0 μL。PCR反应程序参照TB Green Premix Ex Taq Ⅱ试剂说明书进行,每个反应重复3次。基因相对表达量采用2?ΔΔCt法计算,SPSS 27.0统计分析。 3 结果与分析3.1 远志bZIP基因家族成员的鉴定和蛋白理化性质分析基于远志全长转录组数据库,共筛选得到63个注释为bZIP基因的序列ID,进一步分析后获得39个包含完整ORF的序列。整理ORF差异位点并合并重复,最终得到27个全长bZIP转录因子,编号PtbZIP1~PtbZIP27(表1)。该转录因子的氨基酸个数143~846,相对分子质量介于16 201.52~92 932.3,等电点4.59~9.69。除PtbZIP1和PtbZIP22的不稳定指数小于40,系稳定蛋白质外,其余PtbZIP均为不稳定蛋白。bZIP基因家族脂肪系数介于48.31~92.66,所有bZIP蛋白的平均亲水性数值是负值,为亲水性蛋白。图片3.2 远志bZIP基因家族成员的二级结构、信号肽、跨膜结构及亚细胞定位分析二级结构分析结果(表2)表明,远志bZIP家族蛋白均具有α螺旋、延伸链、β转角和无规卷曲,主要由α螺旋和无规卷曲构成,延伸链和β-折叠所占比例较小,散布于整个蛋白中。SignalP-5.0和TMHMM在线分析结果一致,所有远志bZIP蛋白信号肽分值都低于0.5,说明其均无信号肽,不属于分泌蛋白。跨膜结构域分析则显示,仅PtbZIP9和PtbZIP13有跨膜结构域。亚细胞定位结果表明,远志bZIP家族成员主要定位在细胞核。图片3.3 远志bZIP基因家族成员系统进化分析利用MEGA7.0构建远志与拟南芥bZIP转录因子家族系统进化树。结果表明,27个PtbZIP蛋白分为A、B、C、D、F、G、I、S 8个组,没有bZIP蛋白分到E和K组中。其中G是最大的1个亚组,含有PtbZIP家族成员共8个,占总数的29.63%;A、F、I和S组均含3个PtbZIP家族成员,B组含2个PtbZIP家族成员,C组含1个PtbZIP家族成员,D组含4个PtbZIP家族成员(图1)。图片3.4 远志bZIP基因家族成员蛋白结构域分析BRLZ、MFMR和DOG1为bZIP蛋白中的常见结构域,BRLZ参与调控果生炭疽菌的营养生长,MFMR涉及蛋白与蛋白之间的相互作用,DOG1则与种子休眠相关[21-22]。远志bZIP的结构域分析结果表明:10个蛋白存在BRLZ结构域,9个蛋白存在MFMR结构,6个蛋白存在DOG1结构域(图2)。PtbZIP3和PtbZIP13含有大小相近的CCDC 158 superfamily,PtbZIP26、PtbZIP21和PtbZIP5则均含有BRLZ、MFMR及homeobox结构,结合进化树结果可知PtbZIP3和PtbZIP13聚在一起,PtbZIP26、PtbZIP21和PtbZIP5三者亲缘关系较近。图片3.5 远志bZIP基因家族成员保守基序分析利用MEME对远志27个bZIP蛋白序列进行保守基序分析的结果显示,不同bZIP转录因子基因包含的保守元件数量及种类存在差异,其中bZIP14基因包含的保守元件数量最少(2个),bZIP18/25基因包含的保守元件数量最多(11个),说明bZIP成员具有功能冗余现象,也具有功能差异性(图3)。图片bZIP蛋白结合位点序列分析结果表明,bZIP转录因子的每个重复结构域约为65 aa,均含有1个保守的bZIP结构域,其中N端一般具有高度保守的N-X7-R蛋白基序和碱性亮氨酸区域(图4)。图片3.6 远志bZIP基因家族成员密码子偏好性分析密码子可用来推断基因组内部或基因组之间的进化关系,而不同种类或同一种类的基因对密码子使用有不同的偏好模式[23]。由bZIP基因家族中的27条核苷酸序列中密码子GC的总含量(GC)以及同义密码子第1位(GC1s)、第2位(GC2s)、第3位的(GC3s)的GC含量分析结果可知:27条PtbZIP基因序列的GC1s、GC2s和GC3s的均值分别为52.24%、44.90%和40.93%,不同位置的GC含量存在差异;它们的GC平均值为46.11%,小于50%,表明其更偏向于A或U结尾的密码子[24](表3)。图片有效密码子(effective number of codon,ENC)反映了密码子偏离随机选择的结果,它是对同义密码子非均衡使用偏好程度的一个重要指标[25],ENC数值一般在20~61范围内,当ENC>35则表示密码子偏好性较弱。密码子适应指数(codon adaption index,CAI)是指编码该蛋白的所有密码子相对于这条基因都使用最优密码子的情况下的适应系数[24]。由表3可知,远志bZIP家族成员的ENC数值为43.088~57.195个,平均值为51.13个,密码子偏好性较弱。CBI值较低说明其外源基因在目的宿主中表达较弱。CAI值较低,则说明其适应性较弱。3.7 远志bZIP基因家族成员蛋白互作网络分析为深入了解远志bZIP蛋白的潜在功能和家族成员之间的相互作用,利用STRING软件,基于拟南芥数据库,对远志的27个bZIPs蛋白进行了互作网络分析。由图5可知,调控网络中共有27个节点(代表bZIPs蛋白),104条边(代表蛋白质之间的相互作用),表明远志的bZIPs蛋白存在多种互作现象,且26个bZIPs成员之间存在潜在的互作关系,为进一步验证远志bZIP的功能提供了重要依据。图片3.8 远志bZIP基因家族成员不同组织表达量热图和验证根据远志转录组数据,对27个PtbZIP基因在远志根、茎、叶中的FPKM差异表达数据进行了双向聚类分析。通过表达量热图分析可知,绝大部分基因的表达不恒定,在不同组织具有相对较高的表达量,根、茎和叶中表达量较高的基因数分别为23、2和2。PtbZIP4/15在叶中的表达量最高,茎和根次之;PtbZIP8/24在茎中的表达量最高,叶和根次之;剩下23个除PtbZIP1/17的表达量为根>叶>茎,其余表达模式为根>茎>叶(图6-A)。基于RT-qPCR验证转录组数据结果显示,PtbZIP26、PtbZIP27在根中的表达量最高,茎、叶次之,与转录组结果一致(图6-B)。图片3.9 PtbZIP26不同处理下的表达模式为了探究bZIP家族基因在远志不同处理条件下的表达模式,以PtbZIP26为代表,对其进行了激素和干旱、盐胁迫处理条件下的表达模式分析。结果发现,以0 h为空白对照(CK),PtbZIP26的表达量在ABA处理6 h内迅速上升,在24 h达到峰值;CTS处理分别持续上调至峰值为CK的5.3倍(24 h)后逐渐下调(图7-A)。PEG处理6 h迅速下降后又随着处理时间增加缓慢恢复上调,NaCl处理6 h后上调明显(图7-B)。 图片4 讨论bZIP基因家族在植物中广泛分布,参与植物的多个生长过程,如生长发育、应激反应以及次生代谢物的生物合成[26]。现阶段,bZIP基因家族已在多个物种有过相关的鉴定和研究,使得对bZIP的生物功能了解更透彻。本实验基于远志三代全长转录组数据库,找到39个bZIP isoforms,通过完整开放阅读框与BlastX分析找出具有完整ORF的基因,去除重复的isoforms,筛选并鉴定得到27个PtbZIP基因家族成员。理化性质分析显示,27个成员均为亲水性蛋白,且除PtbZIP1和PtbZIP22外均为不稳定蛋白;理论等电点小于7的蛋白有16个,属酸性蛋白,其余均为碱性蛋白。PtbZIP蛋白信号肽分值都低于0.5,说明其均无信号肽,信号肽是分泌蛋白的决定因子,推测PtbZIP蛋白不属于分泌蛋白。亚细胞定位结果显示,远志bZIP蛋白主要定位于细胞核,这与转录因子主要在细胞核中发挥作用一致。PtbZIP家族成员的蛋白二级结构也有明显的特点,主要有α-螺旋、无规卷曲。系统进化分析显示,27个PtbZIP蛋白分为A、B、C、D、F、G、I、S 8个组,其中含有8个PtbZIP家族成员的G亚组系最大亚组。PtbZIP11/18/25与拟南芥At1g32150.1、At2g35530.1高度同源,且包含的保守元件数量最多,推测PtbZIP11/18/25可能在远志干旱应答的分子机制中起重要作用[27]。研究表明,A类别的大多数功能信息提示在ABA或应激信号中的作用,PtbZIP6/12/16被分在A组,推测该基因可能参与到远志ABA信号转导途径[28]。S类别是拟南芥最大的bZIP类别之一,在胁迫处理后也被转录激活或在花的特定部分特异表达。研究证实,拟南芥bZIP家族中的S类别的基因在响应干旱有重要作用,本研究中共有3个PtbZIP基因被分到S类别下,其中PtbZIP15在叶中表达量高,PtbZIP24在茎中表达量高,可能参与调控远志对干旱的响应。同时,27个PtbZIP基因家族成员的蛋白二级结构预测结果十分相似,但序列间同源性相对较低,表明PtbZIP基因可能在远志生长发育方面发挥广泛的生物功能。表达模式分析发现,大部分PtbZIP在根中表达最高,qPCR结果验证与转录组数据一致,推测它们主要在远志地下部分发挥作用。植物中转录因子的表达与激素密切相关,研究发现葡萄VvLysM8和VvLysM9在壳聚糖处理12 h、脱落酸处理3 h时相对表达量最高[14]。马铃薯StHXK家族基因在ABA诱导下表达均显著上调,且在10%PEG胁迫处理下也呈不同程度的上调表达[29]。陆地棉GhKIN基因家族的鉴定和分析发现,干旱和盐胁迫处理后GhKIN14和GhKIN27表达出现下调,而GhKIN18等在一定时间点表现为表达上调[30]。本研究选择一个在根中高表达的PtbZIP26基因,通过不同激素、胁迫处理探讨了其是否受到相关激素和胁迫调控,结表明激素处理(ABA和CTS)远志幼苗后,PtbZIP26表达水平显著提高;同时,盐胁迫和干旱胁迫处理也可诱导PtbZIP26基因的表达发生改变且随胁迫时间的变化呈现出差异性,说明PtbZIP26可能通过不同信号通路参与远志应对逆境胁迫的表达,具体作用机制有待深入研究。本实验基于远志三代转录组数据,以远志bZIP基因家族为研究对象,对其家族成员进行鉴定和生物信息学预测分析,明确了相关结构特点与进化特征,进一步通过qPCR分析其在不同组织、不同处理下的表达模式,为探究PtbZIP参与生长发育、代谢过程及非生物胁迫的调控机制提供参考依据,为后期的基因功能研究奠定了基础。
在后基因体时代,基因芯片 (microarray) 的出现让研究人员得以宏观的视野来探讨分子机转。在许多努力和资源投入到寻找新的疾病基因后,许多单基因疾病已成功地找出致病基因。然而,在复杂疾病 (例如高血压、糖尿病及一些常见癌症) 的研究上,收获却不如期待中的丰富。大多数复杂疾病的研究中都可找出分布在不同染色体上的致病基因,但其与疾病仅有小至中等的连结 (linkage) 或关联性 (association),且只有极少数的致病基因能在大量人口资料中,仍对疾病的连结或关联性具有显着性。目前从复杂疾病研究找到的致病基因,大多数在跨研究的报告中皆不具重现性。 复杂疾病具异质性、多源性以肥胖为例,在2004年Dr. Perusse1的研究发现:与人类肥胖相关的113个候选基因 (candidate gene) 在50个全基因扫描研究中,仅有18个基因在五个以上的研究提出一致的正面相关报导。另外,2005年Dr. Agarwal2 的评论提到 (如图一所示),25个高血压基因在不同的连结或关联性研究中,有9个基因在连结性研究中负面相关的报导多于正面相关的报导。而25个基因中,多数在关联性研究中正面相关和负面相关的报导不相上下。 http://img.dxycdn.com/trademd/upload/userfiles/image/2012/12/A1354777030_small.jpg图1:2005年Dr. Agarwal 的评论中针对25个高血压基因在不同的连结或关联性研究中的统计报导 文献中将复杂疾病的致病基因在跨研究间缺乏重复性的现象,归纳出了几点解释。其中一个最广为接受的看法是这些多因子疾病的异质性 (heterogeneous)。另外,因在不同研究中,对各种表型 (phenotype,如血压、血糖) 定义上的不同和量测的不精确、对环境危险或保固因子 (如抽烟量,对污染物的摄取量) 的不同暴露程度以及不同人口之间基因背景的差异等因素,皆会遮蔽、加强或改变基因的作用并造成不同程度的疾病外显率 (penetrance)。 简而言之,由于复杂疾病患者病因的多源性,稀释了任何一个基因变异的效果。所以,当我们将许多病患集中在一起,试图比较他们的基因和正常人有何不同时可能会发现不同的致病基因,甚至亦会发现跟疾病无关而是与病患其他特性相关的基因。 生物路径丛 (Pathway Clster) 概念目前在复杂疾病的研究上,一般以使用类似的表型以减少样本间的异质性。然而,表型的同质化并不等于基因型的同质化。再者,一个疾病可能只是多种表型类似,但起源(基因)不同的病征组合。这个概念虽曾在文献中被提出过,但科学家所使用的简化表型方法并不尽理想。譬如在精神疾病领域,许多学者提出 ”endophenotype”,也就是「内在生物表型」这个概念。但他们所提出的操作方法,仅只是简单化(或减化)表型,譬如:以解剖学、影像学,或症兆定义上来减化,而没有着眼在减化「参与病征发展的生化路径」上。 这个问题的主要瓶颈在于科学家对于疾病发展的机制还不够了解。因此,中研院潘文涵教授3 提出以下建议:在现今大量产生的基因表现数据上,运用「数据探勘 (data mining)」的方法,进行群组分析 (cluster analysis);将这些资料分成若干个群组内相关,但群组间不相关的多个群组,每一个群组可能代表一两个少数源头基因、和一些他的下游基因的表现状态。所得群组同构型高且接近病原的潜在基因,因此可视为「生物路径丛」的指针。
看了一个招标新闻,中国烟草总公司1022万采购仪器,主要仪器如下: 项目A:超高分辨率激光共聚焦显微镜1套;正置显微镜1套;倒置显微镜1套;切片机1套;偏光体视显微镜1套。 项目B:超低温冰箱15台。 项目C: 生物分析仪1台。 项目D: 基因芯片分析系统一套。这些仪器应该是用于生命科学研究的吧,用在烟草基因研究,烟草基因研究主要是为了啥?
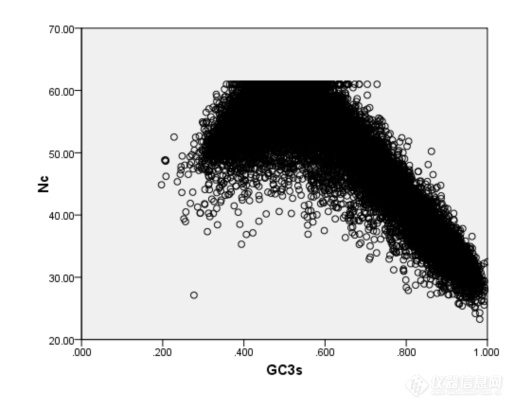
[align=center]短柄草全基因组密码子用法分析分析[/align]摘要:本研究运用CodonW程序分析了短柄草全基因组的密码子使用特性,并且通过对应分析探讨了若干重要因子对短柄草全基因组序列密码子用法的影响。结果表明短柄草基因组存在高[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]含量和低[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]含量的基因,它们在密码子使用上差异较大。Nc-plot曲线表明基因组的密码子组成受到碱基组成的影响;对应分析显示,在DNA水平上发生的核苷酸突变可能是造成短柄草基因组密码子使用偏好的主要因素;同时,基因长度和蛋白质疏水性对密码子的使用也存在一定偏性,但影响程度不大。确定了UUC等27个以G或C碱基结尾的密码子为“最优密码子”,研究结果可为短柄草基因的鉴定、表达、结构、功能等的深入研究提供参考。关键词:同义密码子偏好性,短柄草基因组,对应分析近年来,随着分子生物学的快速发展,许多小基因组的低等生物和高等模式生物的全基因组序列均被测定,为利用生物信息学方法挖掘海量基因组数据提供了便利。密码子是生物体内遗传信息传递的基本环节,是核酸携带信息和蛋白质携带信息间对应的基本规则。在长期进化过程中,任一物种的基因都会逐渐适应宿主的基因组环境,而形成特定的且符合宿主基因组的密码子用法,因此不同生物具有不同的密码子使用模式。以生物基因组数据为基础,研究其密码子使用模式,为深入研究基因的结构、功能和基因组进化,以及指导基因转化等具有重要意义。密码子具有简并性,生物在同义密码子的使用上并不是完全随机的,而是具有一定的偏向性,对有的密码子使用频率高,有的使用频率低,甚至避免使用,这种不均衡使用密码子的现象普遍存在于原核和真核生物中。早在20世纪70年代,人们在研究基因的异源表达时,就已经意识到密码子偏性的重要性[1],随着不同生物基因组数据的获得和各种数据库的构建,更多的研究者对密码子偏性的研究产生了浓厚的兴趣,尤其在分子进化,翻译调控等研究领域,通过对不同物种的密码子使用偏性的大量研究[2~4],发现不同物种的基因在密码子使用上存在着明显的偏性。 短柄草是一种广泛分布于温带地区的禾本科植物,与小麦,大麦和燕麦同属早熟禾亚科,原产于非洲北部,欧洲南部和亚洲中部,包含约10个亚种。该植物为一年生,自花授粉,植株高度15~20cm,生育期70~80d,柄草植株较小,适应性强,不象种植水稻那样需要严格的生长条件。生育期短,籽粒产量较高,一年可以繁殖4~5代,繁殖系数达140左右。未成熟胚和成熟胚愈伤组织诱导率高,农杆菌介导和基因枪介导的转化体系已经建立,胚性愈伤组织分化率90%以上,转化效率最高可达55%左右。基因组小,染色体少,DNA重复序列低,获得突变体容易,突变性状容易显现,具备了模式植物的所有基本特征。加之短柄草基因组序列与黑草麦,小麦,大麦等早熟禾亚科植物高度相似,很多重要农艺性状与温带禾草类植物相似,如株型,穗型,粒型,抗逆性,生长习性和病原菌等,其中麦类作物白粉病菌,条锈病菌和稻类作物瘟病菌都可侵染短柄草植株,引起相应症状[7]。其籽粒不含高分子量麦谷蛋白亚基,低分子量麦谷蛋白亚基也很少,并与小麦一样具有二倍体,四倍体和六倍体,因此短柄草是小麦等基因组庞大的重要农作物理想的模式植物,借此来获得目前小麦等早熟禾类植物中尚缺少的遗传信息和基因共线区,进而对小麦等重要植物进行基因定位,克隆,突变,测序和功能等方面的研究[8]。 目前,在短柄草的生物学、细胞学和遗传学特性方面开展了大量研究,并且其全基因组测序也基本完成[9],为深入研究其密码子用法提供了便利。因此本研究将以短柄草全基因组序列为基础,分析其基因的密码子用法特性和影响密码子使用的因素等,其研究结果将对指导转基因及对基因进行特定分子改造,提高其在短柄草中的表达效率和完善基因预测软件,提高基因预测和基因组注释准确性等均具有重要的参考价值,同时也为深入开展基因结构和功能,分子进化等研究提供理论基础。1.实验材料与方法1.1材料 短柄草全基因组DNA序列来源于短柄草官方数据库(http://www.brachypodium.org/node/8),根据基因组序列的注释信息,获得蛋白编码基因序列,为了减少长度较短的基因变异带来的样本误差,根据国际惯例,去除小于300bp的基因,去除中间不表达的密码子,终止密码子。编写程序提取剩下的蛋白编码基因的CDS(coding sequence)序列。1.2方法用codonw软件计算短柄草全基因组的密码子用法相关参数,主要包括有效密码子数(Effective Number of Codon,ENC)、基因的G+C含量([url=https://insevent.instrument.com.cn/t/Mp]gc[/url]%)、[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]3s%、相对同义密码子使用度(relative synonymous codon usage,RSCU)、氨基酸组分指数(平均亲水性值(gravy))、基因长度即氨基酸数(L_aa)。其中,有效密码子数(Effective Number of Codon,ENC)描述密码子使用偏离随机选择的程度,能反映密码子家族中同义密码子的非均衡性的偏好;其取值范围在20到61之间,即如果每种氨基酸只使用一种密码子则有效密码子数为20,如果各种同义密码子的使用机会完全均等,则有效密码子数为61,数值越小偏性越强。此值是以描述密码子使用偏离随机选择的程度,能反映密码子家族中同义密码子的非均衡性的偏好。基因密码子偏爱程度越大,ENC值越小。RSCU是指对于某种特定的密码子在编码对应氨基酸的同义密码子间的相对频率;[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]3s%表示同义密码子第三位碱基的G+C的含量。为进一步了解该家族基因密码子使用特征和影响密码子使用的因素,对7个基因的相对同义密码子使用度进行了对应性分析(correspondence of analysis,COA)。2 结果与分析2.1 基因的碱基组成对密码子使用的影响图一 短柄草基因NC值散点图[img=,515,409]https://ng1.17img.cn/bbsfiles/images/2019/10/201910311236371230_3093_3295053_3.png!w515x409.jpg[/img]2.2短柄草基因密码子使用特性的对应性分析[img=,690,535]https://ng1.17img.cn/bbsfiles/images/2019/10/201910311237226440_1452_3295053_3.png!w690x535.jpg[/img][img=,690,534]https://ng1.17img.cn/bbsfiles/images/2019/10/201910311237233450_935_3295053_3.png!w690x534.jpg[/img]2.3 确定最优密码子Phe UUU 0.05 (323) 1.23 (19733) Ser UCU 0.22 (990) 1.60 (23834) UUC* 1.95 (13527) 0.77 (12294) UCC* 2.55 (11715) 0.64 (9499) Leu UUA 0.02 ( 93) 0.83 (11755) UCA 0.14 (629) 1.52 (22651) UUG 0.16 (1003) 1.37 (19558) UCG* 1.53 (7023) 0.35 (5159) CUU 0.14 (847) 1.55 (21987) Pro CCU 0.22 (1306) 1.57 (17584) CUC* 3.38 (20676) 0.61 (8661) CCC* 1.35 (7940) 0.47 (5299) CUA 0.07 (452) 0.70 (9983) CCA 0.20 (1184) 1.62 (18078) CUG* 2.23 (13637) 0.94 (13401) CCG* 2.22 (13058) 0.34 (3792) Ile AUU 0.12 (398) 1.41 (21216) Thr ACU 0.10 (401) 1.46 (16515) AUC* 2.76 (9124) 0.70 (10557) ACC* 1.75 (7291) 0.66 (7397) AUA 0.12 (380) 0.89 (13461) ACA 0.12 (509) 1.56 (17636) Met AUG 1.00 (8512) 1.00 (20892) ACG* 2.03 (8478) 0.32 (3563) Val GUU 0.10 (693) 1.67 (23852) Ala [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]U 0.14 (1914) 1.65 (26184) GUC* 1.71 (12491) 0.63 (9025) [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]C* 1.98 (27398) 0.58 (9131) GUA 0.05 (349) 0.75 (10713) [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]A 0.13 (1802) 1.48 (23459) GUG* 2.14 (15605) 0.95 (13562) [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]G* 1.75 (24170) 0.29 (4678) Tyr UAU 0.05 (229) 1.28 (14480) Cys UGU 0.06 (194) 1.10 (9360) UAC* 1.95 (8126) 0.72 (8075) U[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]* 1.94 (6645) 0.90 (7595) TER UAA 0.42 (172) 0.82 (335) TER UGA 1.63 (665) 1.30 (530) UAG 0.94 (384) 0.87 (356) Trp UGG 1.00 (4992) 1.00 (10053) His CAU 0.15 (598) 1.42 (16785) Arg CGU 0.16 (750) 0.85 (6945) CAC* 1.85 (7568) 0.58 (6825) C[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]* 2.75 (12565) 0.49 (4043) Gln CAA 0.15 (627) 1.05 (20215) CGA 0.11 (500) 0.64 (5273) CAG* 1.85 (7975) 0.95 (18259) CGG* 1.92 (8761) 0.55 (4527) Asn AAU 0.12 (465) 1.31 (26650) Ser AGU 0.05 (235) 1.13 (16754) AAC* 1.88 (7141) 0.69 (13985) A[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]* 1.52 (7002) 0.77 (11441) Lys AAA 0.11 (552) 0.98 (27077) Arg AGA 0.10 (445) 1.94 (15854) AAG* 1.89 (9406) 1.02 (28423) AGG 0.96 (4387) 1.53 (12516) Asp GAU 0.15 (1344) 1.44 (39136) Gly GGU 0.11 (882) 1.34 (18423) GAC* 1.85 (16539) 0.56 (15322) G[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]* 2.53 (20795) 0.71 (9826) Glu GAA 0.17 (1437) 1.13 (36292) GGA 0.19 (1522) 1.26 (17423) GAG* 1.83 (15812) 0.87 (27746) GGG* 1.18 (9700) 0.69 (9476) 注:Number of codons in high bias dataset 372333 Number of codons in low bias dataset 915109标注*的密码子是(p 0.01)3 讨论密码子使用偏好是突变偏好、自然选择和遗传漂变等共同作用的结果,与碱基组成、翻译选择压力、基因表达水平、基因长度、蛋白质氨基酸组成、碱基突变频率和模式、mRNA二级结构稳定性等很多因素有关[17]。张晓峰[18]等研究表明,单子叶植物基因组的[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]含量在同义密码子使用偏性的产生过程中起着决定性的作用,同义密码子使用偏性强烈的基因往往偏爱使用C或G结尾的密码子,且第三位密码子突变往往是密码子偏好性发生变化的决定原因。短柄草基因密码子使用模式的调查表明其中有高含量的[url=https://insevent.instrument.com.cn/t/Mp]gc[/url],并且[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]3的含量高于[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]1和[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]2。这表明相对于以A和T结尾的密码子而言,这些基因偏好于使用以G或C结尾的密码子。从原核生物到真核生物的基因中,密码子使用偏好是一个被广泛研究的重要进化现象。研究发现,许多因素,比如碱基组成,基因表达水平,蛋白质疏水性等影响着密码子的使用。为了解释密码子使用偏好的起因,也有许多假设被提了出来。其中被广为接受理论是“选择——突变——漂移”模型。该模型认为在对偏好密码子的选择和通过突变-漂移对非偏好密码子的保留之间,同义密码子的使用偏性存在一种平衡。本文的研究结果显示,[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]3s值与ENC值密切相关,并且基因也位于第一轴线,揭示了碱基组成是影响短柄草基因组中的密码子使用偏好的主要因素。碱基组成是影响短柄草基因密码子使用的主要因素,基因长度和蛋白质的疏水性在短柄草基因密码子使用中也起到了一定的作用,相似的结果在水稻、小麦中被发现[15,19]。本研究发现,在基因长度和[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]之间存在很强的负相关性。这表明,高[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]含量的基因越短,密码子偏好就越大。可能的原因是富含AT基因的翻译效率比富含[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]基因的翻译效率更高,这种效率的差异对长的基因更为重要。通常,全基因组的基因表达值在许多多细胞真核生物中并不能得到,特别是基因表达水平在不同的组织和不同发育阶段不一样时。因此,要定量相当困难。在短柄草基因组中,目前还缺少相当数量的基因表达的准确数据。另外,我们发现[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]含量特别是在第三个碱基位置的[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]含量较大的影响着密码子的偏好时,暗示着碱基突变可能是重要因素,同时,碱基突变又受控于翻译选择。所以,尽管基因表达水平影响着密码子的使用,但这影响还是远远小于核苷酸组成对密码子使用的影响。因此,我们没有进一步分析基因表达的影响。通过优化密码子,提高外源基因在微生物、植物、动物中的表达已有不少成功报道,而确定最优密码子可为合理有效进行密码子改造提供可靠信息。本文确定了UUC等27个密码子为短柄草全基因组的最优密码子。分析结果可为指导转基因及对基因进行特定分子改造,提高其在短柄草中的表达效率和完善基因预测软件,提高基因预测和基因组注释准确性等提供重要的参考价值。参考文献[1] Stanley D,Farnden K J F, MacRae E A. Plant a-amylases:Func-tions and roles in carbohydrate metabolism[J]. Biologia,Bratislava,2005.60(suppl l6):65-71[2] Smith AM. Zeeman SC, Smith S M. Starch degradation[J]. Annu Rev Plant Biol,2005,56(25):73-98[3] Asatsuma S, Sawada C, Itoh K et al. Involvement of α-amylase I-1 in starch degradation in rice chloroplasts[J]. Plant Cell Physiol,2005,4:858-869[4] Kaplan F, Guy C L. β-amylase induction and the protective role of maltose during temperature shock[J]. Plant Physiol, 2004, 1:1674-1684 [5] Kaplan F,Guy C L. RNA interference of Arabidopsis beta-amylase 8 prevents maitose accumulation upon cold shock and increases sensitivity of PSII photochem-ical efficiency to freezing stress[J]. Plant J.2005,44(13):730-743[6] Joho Mundy, Anders Brandt. Messenger RNAs from the Scutellum and Aleurone of Germinating Barley Encode (lm3,14)--D-Glucanase, a-Amylase and Carboxypeptidase[J]. Plant Physiol, 1985,79(5):867-871 [7] 言普,李桂双.高压对水稻种子细胞膜透性和淀粉酶活性的影响[J]. 浙江大学学报(农业与生命科学版),2007,33(5):174-179[8] Monica M, Sanwo and Darleen A. DeMason. Characteristics of a-Amylase during Germination of Two High-Sugar Sweet Corn Cultivars of Zea mays L[J]. Plant Physiol, 1992,99(8):1184-1192[9] Goldman N , Yang Z. A codon based model of nucleotide substitution for protein coding DNA sequences[J]. Molecular Biology and Evolution,1994,11(9):725-736[10] Schmidt W. Phylogeny reconstruction for protein sequences based on amino acid properties[J]. Mol Evol,1995,41(8) :522-530[11] 时成波, 吕安国.改造稀有密码子提高SEA蛋白表达量[J]. 生物工程学报,2002,18(4):477-480[12] Ghosh T C , Gupta S K, Majumdar S. Studies on codon usage in Entamoeba histolytica[J]. Int J Parasitol,2000,30(6): 715-722[13] Musto H, Cruveiller S. Translational selection on codon usage in Xenopus laevis[J].Molecular Biology and Evolution,2001,18(9):1703-1707[14] 廖登群,张洪亮等. 水稻(Oryza sativa L.)a-淀粉酶基因的进化及组织表达模式[J]. 中国农业大学学报,2009,14(5):1-11[15]刘汉梅,何瑞. 玉米密码子用法分析[J]. 核农学报,2008,22(2):141-147[16] Jia M, Luo L. The relation between Mrna folding and protein structure[J]. Biophys Res Commum, 2006,343(4):177-182[17] 赵耀,刘汉梅. 玉米waxy基因密码子偏好性分析[J]. 玉米科学,2008,16(2):16-21 [18] Wang H C,Hickey D A. Rapid divergence of codon usage patterns within the rice genome[J].BMC Evol Biol,2007,15(8):347-356
METHODS 25, 402–408 (2001)Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2-△△CT MethodKenneth J. Livak* and Thomas D. Schmittgen¶,1*Applied Biosystems, Foster City, California 94404; and ¶ Department of Pharmaceutical Sciences, College of Pharmacy,Washington State University, Pullman, Washington 99164-6534原文下载摘要:现在最常用的两种分析实时定量PCR 实验数据的方法是绝对定量和相对定量。绝对定量通过标准曲线计算起始模板的拷贝数;相对定量方法则是比较经过处理的样品和未经处理的样品目标转录本之间的表达差异。2-△△CT方法是实时定量PCR 实验中分析基因表达相对变化的一种简便方法,即相对定量的一种简便方法。本文介绍了该方法的推导,假设及其应用。另外,在本文中我们还介绍了两种2-△△CT衍生方法的推导和应用,它们在实时定量 PCR 数据分析中可能会被用到。关键词:反转录PCR 定量PCR 相对定量 实时PCR Taqman反转录 PCR (RT-PCR )是基因表达定量非常有用的一种方法(1 - 3 )。实时PCR 技术和RT-PCR 的结合产生了反转录定量 PCR 技术(4 ,5 )。实时定量 PCR 的数据分析方法有两种:绝对定量和相对定量。绝对定量一般通过定量标准曲线来确定我们所感兴趣的转录本的拷贝数;相对定量方法则是用来确定经过不同处理的样品目标转录本之间的表达差异或是目标转录本在不同时相的表达差异。绝对定量通常在需要确定转录本绝对拷贝数的条件下使用。通过实时 PCR 进行绝对定量已有多篇报道(6 - 9 ),包括已发表的两篇研究论文(10,11 )。在有些情况下,并不需要对转录本进行绝对定量,只需要给出相对基因表达差异即可。显然,我们说 X 基因在经过某种处理後表达量增加 2.5 倍比说该基因的表达从1000 拷贝/ 细胞增加到2500 拷贝/ 细胞更加直观。用实时PCR 对基因表达进行相对定量分析需要特殊的公式、假设以及对这些假设的验证。2-△△CT方法可用于定量PCR 实验来计算基因表达的相对变化:2-△△CT公式的推导,以及实验设计,有效性评估在Applied Biosystems User Bulletin No.2(P/N4303859)中有介绍。用2-△△CT方法分析基因表达数据在文献中也有报道(5,6)。本文介绍了该方法的推导、假设以及应用。另外,本文还介绍了2-△△CT两种衍生方法的推导和应用,它们在实时定量PCR 数据分析中都可能被用到。1. 2-△△CT方法1.1. 2-△△CT方法的推导PCR 指数扩增的公式是:http://www.biomart.cn/upload/asset/2008/11/10/1226054042.jpg这里,Xn 是第 n 个循环後目标分子数,X0 是初始目标分子数,Ex 是目标分子扩增效率,n 是循环数,C T 代表目标扩增产物达到设定阈值所经历的循环数。因此:http://www.biomart.cn/upload/asset/2008/11/10/1226054008.jpgX T 是目标分子达到设定的阈值时的分子数。C T,X 是目标分子扩增达到阈值时的循环数。Kx 是一个常数。对于内参反应而言,也有同样的公式:http://www.biomart.cn/upload/asset/2008/11/10/1226054009.jpg用XT 除以RT 得到:http://www.biomart.cn/upload/asset/2008/11/10/1226054010.jpg对于使用 Taqman 探针的实时扩增而言, XT 和 RT 的值由一系列因素决定:包括探针所带的荧光报导基团、探针序列对探针荧光特性的影响、探针的水解效率和纯度以及荧光阈值的设定。因此常数K 并不一定等于1 。假设目标序列与内参序列扩增效率相同:http://www.biomart.cn/upload/asset/2008/11/10/1226054011.jpg http://www.biomart.cn/upload/asset/2008/11/10/1226054018.jpg或:http://www.biomart.cn/upload/asset/2008/11/10/1226054019.jpgXN 代表经过均一化处理过的初始目标分子量;△CT表示目标基因和内标基因CT值的差异(CT,X-CT,R )整理上式得:http://www.biomart.cn/upload/asset/2008/11/10/1226054020.jpg最后用任一样本q 的XN 除以参照因子(calibrator, cb)的XN得到:http://www.biomart.cn/upload/asset/2008/11/10/1226054021.jpg在这里http://www.biomart.cn/upload/asset/2008/11/10/1226054023.jpg对于一个少于150bp 的扩增片断而言,如果 Mg2+ 浓度、引物都进行了适当的优化,扩增效率接近于1 。因此目标序列的量通过内均一化处理之后相对于参照因子而言就是http://tong.dxy.cn/upload/asset/2008/11/10/1226054022.jpg
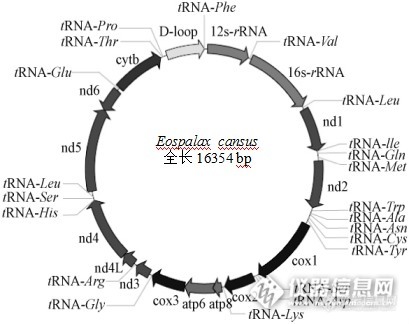
[b][/b][align=center]鼢鼠([i]Eospalax[/i])线粒体基因组测定及注释分析[/align][align=center]西安国联质量检测技术股份有限公司[/align][align=center]安平中心:李瑞[/align][b]摘要【[/b]目的】获得鼢鼠线粒体基因组全序列,为线粒体基因组功能标记及进化生物学等研究提供基础资料。【方法】参考鼹型鼠等动物的线粒体基因组序列,设计出可覆盖鼢鼠线粒体基因组的16对引物,采用[url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url]产物直接测序法测得甘肃鼢鼠线粒体基因组全序列,分析其基因组的特点和基因结构。并结合GenBank中发表的啮齿类动物基因组全序列,探讨啮齿类动物的系统进化关系。【结果】鼢鼠线粒体基因组全长16354bp,其中包括22个tRNA基因、13个蛋白质编码基因、2个rRNA基因和2个D-loop区。碱基组成为33.5%A、24.2 %C、12.3 %G、30.0 %T。【结论】鼢鼠线粒体基因组结构及其信息和其他啮齿类动物的结构一致,线粒体变异保守。研究结果为鼢鼠的低氧适应、系统发育关系等提供了基础资料。[b]关键词 [/b]鼢鼠;线粒体基因组;序列分析 鼢鼠([i]Eospalax[/i])是分布于我国的主要啮齿类动物之一,其体型较小,栖息于洞穴内有挖掘活动,扩散能力强,数量波动大,是生态系统中重要的初级消费者,处于生态系统中的中心位置,草原生态系统中其能流比重很大[sup][/sup]。动物线粒体([color=#333333]Mitochondrion[/color])基因组为双链闭合环状分子[sup][/sup],少数也有线性的,它们具有分子量相对较小、结构简单、缺少重组、母性遗传和进化速率快等特点,已成为动物系统发育与进化、群体遗传学、分子生态学以及疾病机理研究等领域的理想材料[sup][/sup]。甘肃鼢鼠是仅分布于我国西北部的土著物种,其外形似中华鼢鼠,主要分布于甘肃临潭县及其附近地区。目前对线粒体DNA的研究主要在动物分子遗传学、分子生态学、种群遗传结构分析、遗传多样性、物种和品系鉴定、保护遗传学等方面得到了广泛应用[sup][4[/sup][sup],[/sup][sup]5][/sup]1. [b]实验材料和方法[/b]1.1 实验材料鼢鼠:采集于天祝(经度102.84、纬度 37.2)1个群体;鼢鼠解剖采集肝脏及肌肉组织样品,-20℃保存备用。1.2 线粒体DNA的提取用剪刀将肝脏及肌肉材料剪成小块,取0.1cm左右的小块肝脏及肌肉材料,采用常规的SDS/蛋白酶K裂解,酚氯仿提取DNA[sup][/sup],使用琼脂糖凝胶电泳检测其完整性。1.3 引物设计和[url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url]扩增通过Clustal X1.83比对,寻找相对应保守区域位置,用Primer Premier5.0引物设计软件设计引物,并对每条引物进行评价和修改,最终确定16对引物。以所提取的DNA为模板,用16对引物扩增覆盖整个线粒体基因组。利用引物进行[url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url]扩增,反应体系总体积为50μL,其中含有6μL [url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url] buffer、3μL MgCl[sub]2[/sub](1.5mmol)、MgCl[sub]2[/sub],2μL dNTPs (100μL mol)、上下游引物各2μL (0.25μL mol)、Taq DNA聚合酶2μL (1U)、总DNA约为2μL (25ng)、去离子水31μL。反应程序为:94℃预变性4 min,94℃变性50s,48-45℃退1min,72℃延伸1 min 30s,循环30次,之后72℃延伸10min,并根据不同引物的退火温度和扩增反应的实际效果进行优化。取 5 μL [url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url]扩增产物,和2 μL DNA marker 2000,进行1.0%琼脂糖凝胶(1×TBE)5V/电泳,用紫外观察[url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url]产物扩增情况,凝胶成像仪扫描记录结果。1.4 纯化、测序和序列拼接 在[url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url]产物中加入5 U SAP和2 U ExoⅠ,震荡混匀,37℃保温1 h,然后75℃保温15 min以灭活SAP和ExoⅠ酶,纯化好的模板可以在4℃保存24 h或-20℃长期保存。将纯化后的引物送往上海生工生物技术服务有限公司用ABI-3730序列自动分析仪进行双向测序。利用DNASTAR和测序峰图结果分析软件Chromas 2.22校对测序图,DNAMAN拼接序列。得到甘肃鼢鼠线粒体全基因组全序列。2. [b]结果[/b]2.1 鼢鼠线粒体基因组基因定位2.2.1 鼢鼠线粒体2个rRNA的分析哺乳动物线粒体的rRNA具有高度的保守性,它们的位置固定,12S rRNA位于tRNA-phe 和tRNA-Val之间,16S rRNA位于tRNA-Val和 tRNA-Leu之间,12S rRNA起始位置为68,终止位置为1019,长度为952bp,16S rRNA起始位置为1086,终止位置为2651,长度为1566。同时我们比对了鼢鼠和中华鼢鼠的rRNA基因和蛋白质基因,12S rRNA和16S rRNA的相似性分别为91.0%和87.3%,高于蛋白质编码基因之间的相似性。2.2.3 鼢鼠线粒体基因组结构 除NADH脱氢酶亚基6外均在H链上,虽然鼢鼠染色体数目少、染色体大,但与其它哺乳动物线粒体全基因组相比,它的线粒体基因组的结构与其它哺乳动物是十分相似的。甘肃鼢鼠线粒体基因组结构见图1。[align=center][img=,409,324]http://ng1.17img.cn/bbsfiles/images/2017/09/201709081454_02_2904018_3.png[/img][/align]注:ND: NADH脱氢酶亚基(NADH dehydrogenase subunit)、Cox:细胞色素氧化酶亚基(cytochrome oxidase subunit)、Atp:ATP合成酶亚基(ATP synthase F0 subunit)、Cyt b:1个细胞色素b编码基因(cytochrome b)。[align=center][b]图1[/b] 甘肃鼢鼠线粒体基因组结构简图[/align][align=center]Fig.1 The gene organization of [i]Eospalax cansus[/i] mitochondrial genome[/align]3. [b] 讨论[/b] 甘肃鼢鼠线粒体基因组的D-loop区,长度为933bp,比中国地鼠D-loop区(867bp)长。D-loop区对目的基因是不可缺少的,虽然D-loop区不能编码蛋白质但对于遗传信息表达是不可缺少的,在它上面有调控遗传信息表达的核苷酸序列,具有遗传效应的,比如RNA聚合酶结合位点是具有遗传效应的。8只甘肃鼢鼠中有5个单倍型:3只临潭群体共享1个单倍型,2只天祝群体独享单倍型;其余个体均独享单倍型,表明了甘肃鼢鼠线粒体DNA D-loop区碱基变异快、进化快的特性,符合啮齿动物线粒体变异大的现象。随着研究的深入,以线粒体DNA中完整的基因序列或多个基因序列协同而获得遗传信息来探讨物种的系统进化关系,将是以后研究发展的主要方向[sup][/sup]。目前,线粒体DNA已经在许多哺乳类动物的起源进化的研究中取的了重大进展,而对甘肃鼢鼠的起源进化的研究却很少,并且存在着甘肃鼢鼠属于[url=http://baike.baidu.com/view/113192.htm][color=#000000]瞎鼠科[/color][/url]和仓鼠二者之争,因此,为了更好的阐明甘肃鼢鼠的起源,还需要做更多、更深入的研究。
日本 PCR分析 猪牛鱼等基因技术
来自美国麻省理工学院怀海德研究所的研究人员报道,在当前许多各种不同的生物学研究中,用于产生和理解全局基因表达分析数据的常见假设能够导致关于基因活性和细胞行为方面严重缺陷性的结论。相关研究结果刊登在Cell期刊上。怀海德研究所研究员Richard Young说,“表达分析是当代生物学最经常用到的方法之一。因此,我们担心存在缺陷的假设可能影响对很多生物学研究的理解。”今天对基因表达数据的大多数理解都依赖于一种假设:用来分析的所有细胞拥有类似的mRNA总量,其中mRNA大约占细胞RNA中的10%,作为蛋白合成的蓝图发挥作用。然而,一些细胞,包括恶性癌细胞,要比其他细胞产生几倍多的mRNA。传统的全局基因表达分析通常忽略这些差别。Young实验室研究员和论文共同通讯作者Tony Lee说,“我们着重研究了基因表达分析的这种常见性的假设,它潜在影响了很多研究人员。我们提供一种具体的问题例子和一种研究人员能够执行的解决方法。”Young实验室的成员们最近在研究表达高水平c-Myc的癌细胞的基因表达时揭示出这种缺陷。已知c-My是一种基因调节物,在恶性癌细胞中高度表达。当比较表达高水平c-Myc的细胞和表达低水平c-Myc的细胞时,他们吃惊地发现不同的基因表达分析方法能够产生显著性的不同结果。进一步的研究揭示出在含有高水平c-Myc的和低水平c-Myc的细胞中存在显著性的不同,不过这些不同利用常见使用的实验方法和分析方法来掩盖掉。论文共同作者Jakob Lovén说,“我们从不同的基因表达分析方法中观察到的不同结果是令人震惊的,而且导致我们在几种平台上重新研究了这整个过程。我们然后意识到细胞含有类似mRNA水平的常见假设存在严重缺陷,能够导致严重性的误解,特别是对拥有非常不同RNA含量的癌细胞而言,尤其如此。”除了描绘出这种问题之外,研究人员也描述了一种补救方法。通过利用被称作RNA spike-in的人工合成mRNA作为标准对照,他们能够比较实验数据并且能够消除关于细胞RNA总量方面的假设。他们将这种补救方法应用到他们研究的所有三种基因表达分析平台。尽管研究人员相信使用RNA spike-in应当成为全局基因表达分析的新标准,但是理解很多之前的研究时产生的问题可能持续存在。(生物谷Bioon.com)http://www.bioon.com/biology/UploadFiles/201210/2012102722451179.gifdoi: 10.1016/j.cell.2012.10.012PMC:PMID:Revisiting Global Gene Expression AnalysisJakob Lovén, David A. Orlando, Alla A. Sigova, Charles Y. Lin, Peter B. Rahl, Christopher B. Burge, David L. Levens, Tong Ihn Lee, Richard A. YoungGene expression analysis is a widely used and powerful method for investigating the transcriptional behavior of biological systems, for classifying cell states in disease, and for many other purposes. Recent studies indicate that common assumptions currently embedded in experimental and analytical practices can lead to misinterpretation of global gene expression data. We discuss these assumptions and describe solutions that should minimize erroneous interpretation of gene expression data from multiple analysis platforms.
以转基因鲤和非转基因鲤鱼肉(受试物)作为大鼠的饲料基础蛋白成分,从毒性方面开展转基因鲤的食用安全检测分析。急性经口毒性检测结果表明,实验期间,受试动物未见明显的中毒表现,无死亡,尸检中主要脏器亦未见异常,说明转大麻哈鱼生长激素基因鲤食用无毒。将转基因鲤以不同质量分数(10%、5%、2.5%)掺入饲料喂饲大鼠90d,检测结果表明,受试动物活动自如,被毛有光泽,鼻、眼、口腔无异常分泌物。一般毒性检测指标与阴性对照组比较,无显著性差异;末见该受试物对大鼠的血液学指标、血生化指标、尿常规和生化指标、脏器系数、病理组织学指标有不良影响。据此评价转大麻哈鱼生长激素基因鲤在大鼠实验阶段属无毒。
中国科技网讯 据物理学家组织网8月29日(北京时间)报道,美国能源部劳伦斯·利弗莫尔国家实验室(LLNL)研究人员最近开发出一种核酸(DNA和RNA)快速扩增技术,使聚合酶链式反应(PCR)的速度大大加快,可在3分钟内将基因组片段扩增10亿倍,迅速识别出病原菌。疾病快速诊断有望很快成为现实。相关论文发表在最近出版的《分析师》杂志上。 PCR技术能让研究人员把一段DNA或RNA复制上百万副本,然后用于基因组测序、基因分析、遗传病诊断、亲子鉴定、法庭鉴定、确定疾病感染等。该过程一般需要1小时到几天时间。然而,快速诊断、应急反应或传染病监控往往要求PCR技术缩短到几分钟。 领导这项研究的工程师雷金纳德·比尔和同事克服酶动力学和热动力学方面的限制,用多孔材料和绝热薄膜制造出一种设备,实现了极速热循环,能每秒钟加热或制冷45℃,一次热循环不超过2.5秒。比尔特别指出:“这种设备的独特之处还在于,它制冷的速度和加热一样快。” 开发出这种设备后,比尔和同事从10种商用酶中选出了2种,这2种酶的链式反应速度非常快,将一些参数略作调整,就能使反应更快。 他们用一种肠杆菌属的细菌测试了新的PCR设备迅速扩增DNA片段的能力,然后用一段严重急性呼吸道综合征(SARS)DNA片段演示了设备处理威胁公共健康病毒方面的效果。该设备完成对目标DNA30个周期(10亿倍)的PCR扩增,用时仅为2分18秒。 目前,研究小组正在开发一种实时探测设备。按照他们的设想,将来一台PCR仪器就能完成整个测试,从样本到结果只需10分钟。市场对这种设备的需求将是巨大的,除传统的公共卫生和医疗研究领域,一台简单实用的实时PCR设备在养殖、农业以及食品加工行业都非常有用,可用来保障食品安全。(记者 常丽君) 总编辑圈点 随着人类基因组逐渐被破译,一张生命之图将被绘就,我们对人类自身的了解也会迈上新的台阶,很多疾病的病因将被揭开,药物就会设计得更好,治疗方案也能“对因下药”,生活起居、饮食习惯有可能根据基因情况进行调整,人类的整体健康状况将会提高。然而,病来如山倒,为了尽快找到病因,疾病的快速诊断就显得异常重要。而文中提到的技术,可在三分钟内识别病原菌,无疑为很多急症患者的生存争取了宝贵的时间。 《科技日报》(2012-8-30 一版)
1.巴西坚果与转基因大豆事件。美国先锋种子公司将巴西坚果中编码2S albumin蛋白的基因转入大豆中,提高了转基因大豆中的含硫氨基酸。1994年,该公司对该转基因大豆进行食用安全评价时,发现对巴西坚果过敏的人同样会对这种大豆过敏。因此认为,蛋白质2S albumin可能正是主要过敏原,于是立即终止了这项研究计划。但此事后来一度被说成是“转基因大豆引起食物过敏”,作为反对转基因的一个主要事例。 实际上“巴西坚果事件”是研发单位在开展安全评价时发现过敏及时停止的转基因案例,这种转基因大豆也根本没有上市。恰恰说明对转基因植物的安全管理和生物技术育种技术体系具有自我检查和自我调控的能力,能有效地防止转基因食品成为过敏原。 2.普斯泰土豆事件。1998年,据苏格兰Rowett研究所的科学家阿帕得•普斯泰(Arpad Pusztai)称,他在实验中用转雪花莲凝集素基因的马铃薯喂食大鼠,大鼠“体重和器官重量严重减轻,免疫系统受到破坏”。 英国皇家学会(The Royal Society)1999年5月的评审报告指出,普斯泰的实验存在失误和缺陷,主要包含试验设计不科学,试验过程错误百出,试验结果无法重复,因此结果和相应的结论不可信。并且认为,普斯泰在尚未完成实验并且没有发表数据的情况下,就通过媒体向公众传播其结论是非常不负责任的。 3.美国帝王蝶事件。1999年5月,康奈尔大学昆虫学教授洛希(Losey)撰文称,他用拌有转Bt基因抗虫玉米花粉的马利筋杂草叶片饲喂帝王蝶(Monarch butterfly)幼虫,发现这些幼虫生长缓慢,并且死亡率高达44%。洛希认为这一结果表明抗虫转基因作物同样对非目标昆虫产生威胁。 美国环境保护局(EPA)组织昆虫专家对帝王蝶问题展开专题研究。结论认为,该实验是在实验室完成的,并不反映田间情况,且没有提供花粉量数据。评价转基因作物对非靶标昆虫的影响,应以田间实验为准,而不能仅仅依靠实验室数据。2001年10月,洛希研究组又在《PNAS》杂志发表文章称:帝王蝶幼虫经转Bt基因抗虫玉米Bt11 和 Mon810花粉饲喂14到22天对其存活的影响可以忽略不计。 4.墨西哥玉米事件。2001年11月,美国加州大学伯克利分校的微生物生态学家David Chapela和David Quist发表文章,指出在墨西哥南部地区采集的6个玉米品种样本中,发现了一段可启动基因转录的DNA序列——花椰菜花叶病毒(CaMV)“35S启动子”,同时发现与诺华(Novartis)种子公司代号为“Bt11”的转基因抗虫玉米所含“adh1基因”相似的基因序列。绿色和平组织借此消息大肆渲染,说墨西哥玉米已经受到了“基因污染”,甚至指责墨西哥小麦玉米改良中心的基因库也可能受到了“基因污染”。 该文章发表后受到很多科学家的批评,指其实验在方法学上有很多错误。经反复查证,文中所言测出的“CaMV35S启动子”为假阳性,并不能启动基因转录;文中所指在墨西哥地方玉米品种中测出的“adh1基因”是玉米中本来就存在的“adh1-F基因”,与转入“Bt玉米”中的“adh1-S基因”序列并不相同。《Nature》杂志于2002年4月11日刊文,批评该论文结论是“对不可靠实验结果的错误解释”,并在同期申明“该文所提供的证据不足以发表”。 5.中国Bt抗虫棉事件。2003年6月3日,南京环境科学研究所与绿色和平组织在北京召开会议,发布“转Bt基因抗虫棉环境影响研究综合报告”,随后被很多媒体转载刊发,引发国际争论,成为国际上争论转基因抗虫棉安全性的重大事件之一。 国际上的评论是:文章没有经过同行评审,没有说明研究方法,没有生物学统计数据,违反生物学的一般常识,只是按作者的个人意愿断章取义。多国科学家也纷纷发表评论反驳绿色和平组织的观点,认为,抗虫棉不是“无虫棉”,抗虫棉中的Bt基因主要是针对鳞翅目的某些害虫,并不杀死所有害虫,包括盲蝽象、红蜘蛛及甜菜夜蛾。棉农只要采取适当防治措施,如喷洒一般有机磷或菊酯类农药,这些害虫便可得到有效控制,根本谈不上“超级害虫”,更不能说是抗虫棉破坏环境。 6.发生于法国的孟山都转基因玉米事件。2007年,法国分子内分泌学家Seralini及其同事对孟山都公司转抗虫基因玉米的原始实验数据进行统计分析,得出老鼠在食用转基因玉米后受到了一定程度的不良影响。2009年,该研究组再次把欧盟转引的美国孟山都公司的实验数据做了一个粗浅分析,就发表文章“三种转基因玉米品种对哺乳动物健康影响的比较”。文中指出,食用了90天转基因玉米(抗除草剂玉米NK603,抗虫玉米MON810和MON863)的老鼠,与食用转基因玉米不到90天的老鼠,其肝肾生化指标有差异,认为这种差异解释成食用转基因玉米后造成的。 欧洲食品安全局(EFSA,European Food Safety Authority)转基因生物小组对该论文进行了评审,认为该实验结果不是建立在亲自对老鼠进行独立实验的基础之上,文中进行统计分析的数据,是借用来源自孟山都公司之前的实验,而且对数据选择了不合适的、不被同行使用的统计方法作了重新分析。因此结果和结论都是不科学的。来自美国、德国、英国和加拿大的6位毒理学及统计学专家组成同行评议组,对Seralini等人及孟山都公司的研究展开复审和评价,评价结果是:Seralini等人对孟山都公司原始实验数据的重新分析,并没有产生有意义的新数据来表明转基因玉米在3个月的老鼠喂食研究中导致了不良副作用。 7.俄罗斯之声转基因食品事件。2010年4月16日,俄罗斯广播电台“俄罗斯之声”栏目,以《俄罗斯宣称转基因食品是有害的》为题报道了一则新闻。新闻称,由全国基因安全协会和生态与环境问题研究所联合进行的试验证明,转基因生物对哺乳动物是有害的。引用负责该试验的Alexei Surov博士的话说,用转基因大豆喂养的仓鼠第二代成长和性成熟缓慢,第三代失去生育能力。“俄罗斯之声”还称“俄罗斯科学家的结果与法国、澳大利亚的科学家结果一致。当科学家证明转基因玉米是有害的,法国立即禁止了其生产和销售。” 经调查,Alexei Surov博士所在的Severtsov生态与进化研究所并没有任何研究简报或新闻表明Alexei Surov博士曾写过这样的报道,“俄罗斯之声”报道的新闻事件也没有在任何学术期刊上发表过研究论文。至于新闻中提到法国禁止了转基因玉米的生产和销售,事实是法国政府并没有对转基因食品的生产和销售下禁令,而是欧盟已经于2004年5月19日决定允许进口转基因玉米在欧盟境内销售。 8.中国广西迪卡007/008玉米事件。2010年2月,一篇题为《广西抽检男生一半精液异常,传言早已种植转基因玉米》、署名为张宏良的帖子在网络上传播甚广,引发了不少公众对转基因产品的恐慌。文章称,“迄今为
上世纪九十年代,基因疗法首次用于治疗“重度联合免疫缺陷症”(SCID),至今已经进行了两千余例的人体试验。早期临床试验表明,基因疗法在治疗白血病、血友病、地中海贫血、帕金森症、阿尔茨海默病等上效果显着,甚至能够令盲人重获光明。而更多的动物模型试验显示,基因治疗大有根治更多顽疾的可能。2012年5月《科学·转化医学》杂志发表的论文,对逾十年基因治疗受试者的血液样本进行分析,得出结论——“T细胞遗传修饰是一种安全的基因疗法”。这或许能部分消解近年来人们对于基因疗法的过度疑虑。宾夕法尼亚大学佩雷尔曼医学院的布希曼教授(Frederic D. Bushman)为研究基因疗法的长期有效性和安全性,对接受基因治疗的HIV阳性患者进行长期随访。这些患者在1998—2005年间分别接受了一次或数次“T细胞免疫重建”。这种基因治疗是采用传统的逆转录病毒载体,将嵌合抗原受体基因导入患者体内,该嵌合抗原受体能引导机体的免疫系统杀伤HIV感染的细胞。布希曼教授的研究结果表明:患者在接受基因治疗十年后,导入外源基因依然能够发挥治疗效果。数据显示,该基因疗法的半衰期可达16年,这表明该基因疗法在患者体内的有效作用时间可达十余年。说不定,基因疗法真的是未来我们的唯一救星呢。
欢迎刚入门的朋友来看看,老手就别笑话了[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=25784]基因芯片分析的理论与方法[/url]

[img=,351,220]http://ng1.17img.cn/bbsfiles/images/2018/02/201802061354219789_7475_1667_3.jpg!w351x220.jpg[/img]ABI 3130 / ABI 3130XL测序仪的主要性能特点:? 3130xl 采用 16 道毛细管,3130 系统采用 4道毛细管? 24 小时无人监控操作? 仪器设置更方便更简单? 新型自动灌胶系统进行灌胶? 检测池加热器改进了温度控制? 通过 96 孔和 384 孔板自动进样? 3130 POP-7?、POP-6? 和 POP-4? 分离胶? 多色荧光检测? 一种分离胶一种毛细管用于多种应用系统组成美国应用生物系统公司 3130 和 3130xl 基因分析仪由以下组件组成:? 毛细管电泳仪? 计算机工作站:用于仪器控制和数据分析? 软件:用于仪器控制、数据收集和样品文件自动分析? 分析软件包括:- 用于碱基识别的序列分析软件- 用于微卫星、SNP、AFLP 和 LOH 分析的 GeneMapper?/GeneMapper? ID 软件毛细管束提供预组装的 4 根或 16 根内壁无涂层毛细管。毛细管束有多种长度,为多种应用和分析方法提供支持。这些毛细管束在 3130xl 系统上的特定使用寿命为 100 次,在 3130 系统上为 150 次。它们与业界标准的 96 孔和 384 孔板配合使用。分离胶3130 POP-7?、POP-6? 和 POP-4? 三种分离胶(性能优化分离胶)均可以在美国应用生物系统公司的 3130 和 3130xl 基因分析仪上用作分离介质。在每次分析之前,毛细管自动使用新胶进行补充,这些新胶动态覆盖毛细管壁,以消除电渗流。试剂美国应用生物系统公司提供下列用于 3130 系列系统的试剂:? 基因序列分析试剂盒- BigDye? Terminator 荧光标记终止物试剂盒- dGTP BigDye? Terminator 荧光标记终止物试剂盒- BigDye? Primer Cycle 荧光标记引物试剂盒 Sequencing Ready Reaction 试剂盒,M13Rev/-21 M13- dRhodamine Dye Terminator 荧光标记终止物试剂盒? 片段分析试剂盒- Linkage Mapping Set Version 2.5 疾病基因定位克隆试剂盒- GeneScan?-400 HD 荧光标记分子量内标- GeneScan?-500 荧光标记分子量内标- GeneScan?-120 Liz? 荧光标记分子量内标- 其它专用试剂盒样品要求3130 系列系统可以分析由各种样品制备方法制备的多种类型的模板。可以从 96 孔和 384 孔微孔板中自动进样。? 激光 氩离子多波长单模激光器,主要激发波长为:488 和 514.5 nm。? 检测光学系统 美国应用生物系统公司 3130 系统基因分析仪使用专利的激发光和荧光检测光路 来增强荧光信号强度的均一性。这些检测光学系统实时检测来自各泳道毛细管的低本底、全波长光谱数据。毛细管的外径 (od)、内径 (id) 和间距都已进行优化,将因折射引起的信号丢失降至最低。? 电泳电压 最高 可达20 kV? 操作温度范围 18°C-65°C?计算机最低要求- 硬件:Pentium? IV 处理器,2.00 GHz 处理器- 操作系统:Windows XP? Professional Edition- 安装内存 (RAM):1 GB- 硬盘容量:两个 36 GB 硬盘驱动器- 外围设备:CD/RW光盘刻录?运行环境- 温度: 20°C-25°C- 仪器运行时,室温应在 ±2° 范围内波动。- 湿度:40%-60%(无冷凝)? 主电源电压- 220V ±5%- 50-60 Hz ±10%? 电流 最大:15 安培?最大功率 2,000 瓦特(大约)仪器大小尺寸电泳设备:?宽度(门关时):74 cm 宽度(门开时):148.6 cm(左右门同时打开}? 厚度:54.8 cm? 高度:81 cm? 重量:130 kg(大约)
公司现有1台ABI3100 测序仪,成色较新,价格优惠,质量保证,可预约试机。电话:13717963569自动化程度高:提供连续,无监控的操作,自动灌胶,上样,电泳分离,检测及数据分析,可连续运行24小时无需人工干预。样品分析量大:可同时对16个样品进行全自动分析,一天可完成数百个样品的测序或片段分析工作先进的荧光检测系统:采光栅分光,CCD摄像机成像技术,实现多色荧光同时检测应用广泛:除了新基因测序或比较测序工作外,可进行多种片段分析,包括微卫星DNA分析,比较基因型分析,单核苷酸多态性(SNP)研究
从上个世纪八十年代世界上第一例转基因植物诞生以来,转基因的相关研究日益加快,转基因的产品越来越多。据初步统计,关于转基因作物的研究论文2000年为676篇,2009年增加为1301篇,这十年总论文数量为10171篇。相关的新闻报道更是不计其数,其中偶尔有些反对转基因的声音。在生物技术育种二十多年的发展中,千千万万的关于转基因科学研究中,仅有八个所谓的“转基因的安全性问题”事例,其中六个与食用安全相关,两个与生态安全相关。本文目的是通过对这些事例的剖析,看清楚这些事例是否是以科学为基础。同时也让更多的公众真正了解事情的真相。
最近,来自美国加利福尼亚大学圣地亚哥分校、克雷格·文特尔研究院和Illumina公司的科学家对现代基因测序算法进行了改良,只需从一个细菌细胞中提取的DNA(脱氧核糖核酸)就可组装成接近完整的基因组,准确率达到90%,而传统的测序方法至少需要10亿个相同的细胞才能完成。这一突破为那些无法培养的细菌提供了测序方法。研究发表在9月18日的《自然·生物技术》网络版上。 实验室无法培养的细菌范围极广,约占99.9%,从产生抗体和生物燃料的微生物,到人体内的寄生菌。它们的生存条件特殊,比如必须和其他菌种共生,或只能生存在动物皮肤上,因此很难进行人工培养。 论文合著者、文特尔研究院的罗杰·拉斯肯教授10年前曾开发出一种多重置换扩增(MDA)技术,可对实验室无法培养的细菌测序,能恢复70%的基因。其工作原理是对一个细胞的基因片断多次复制,直到其数量相当于10亿个细胞那么多。不过,这种技术却给测序软件带来很多麻烦,它在复制DNA时会出现各种错误,而且并非完全统一放大,有些基因组被复制数千次,有一些却只被复制一两次。但测序算法不能处理这些不一致,而是倾向于舍弃那些只复制了少数次的基因,即使它们对整个基因组来说很关键。 加州大学圣地亚哥分校雅各布工程学院计算机科学教授、现代基因测序技术算法创建人帕维尔·帕夫纳和同事改进了这一方法,保留了那些少量复制的基因片断,并用新方法对一个大肠杆菌测序以检验其精确性,发现它能恢复91%的基因,接近传统的培养细胞水平。这已足够解答许多重要的生物学问题,比如该细菌能产生什么抗体。 人体细菌占体重的约10%,它们有些会造成传染病,但也有的能帮助消化,最近研究还发现,它们能改变人的行为方式,比如引诱人吃更多的东西。新方法也有助于科学家理解细菌行为,研究人体内细菌能产生哪种蛋白质和多肽,这些蛋白质和多肽是细菌之间、细菌和宿主之间互相沟通的工具。 研究小组还用新方法对一种以前未曾测序过的海洋细菌进行了测序,获得了相当完整而且能解释的基因组,掌握了它是如何生存和运动的,该基因组将被存入美国国家卫生研究院的基因银行(GenBank)。研究人员表示还将对更多迄今未知的细菌进行测序。
1、基因芯片综合分析软件ArrayVision 7.0 一种功能强大的商业版基因芯片分析软件,不仅可以进行图像分析,还可以进行数据处理,方便protocol的管理功能强大,商业版正式版:6900美元。 Arraypro 4.0 Media Cybernetics公司的产品,该公司的gelpro, imagepro一直以精确成为同类产品中的佼佼者,相信arraypro也不会差。phoretix™ Array Nonlinear Dynamics公司的基因片综合分析软件。J-express 挪威Bergen大学编写,是一个用JAVA语言写的应用程序,界面清晰漂亮,用来分析微矩阵(microarray)实验获得的基因表达数据,需要下载安装JAVA运行环境JRE1.2后(5.1M)后,才能运行。2、 基因芯片阅读图像分析软件 ScanAlyze 2.44 斯坦福的基因芯片基因芯片阅读软件,进行微矩阵荧光图像分析,包括半自动定义格栅与像素点分析。输出为分隔的文本格式,可很容易地转化为任何数据库。 3、 基因芯片数据分析软件 Cluster 斯坦福的对大量微矩阵数据组进行各种簇(Cluster)分析与其它各种处理的软件。 SAM Significance Analysis of Microarrays 的缩写,微矩阵显著性分析软件,EXCEL软件的插件,由Stanford大学编制。4.基因芯片聚类图形显示 TreeView 1.5 斯坦福开发的用来显示Cluster软件分析的图形化结果。现已和Cluster成为了基因芯片处理的标准软件。FreeView 是基于JAVA语言的系统树生成软件,接收Cluster生成的数据,比Treeview增强了某些功能。 5.基因芯片引物设计 Array Designer 2.00 DNA微矩阵(microarray)软件,批量设计DNA和寡核苷酸引物工具。
基因芯片技术进展及应用 作者:刘炎 [关键词] 基因芯片;核酸探针序列;杂交 1 基因芯片概述 随着人类基因组计划( Human Genome Project)即全部核苷酸测序的即将完成,人类基因组研究的重心逐渐进入后基因组时代( Postgenome Era)向基因的功能及基因的多样性倾斜[1,2]。通过对个体在不同生长发育阶段或不同生理状态下大量基因表达的平行分析,研究相应基因在生物体内的功能,阐明不同层次多基因协同作用的机理,进而在人类重大疾病如癌症、心血管疾病的发病机理、诊断治疗、药物开发等方面的研究发挥巨大的作用。它将大大推动人类结构基因组及功能基因组的各项基因组研究计划。 基因芯片的工作原理与经典的核酸分子杂交方法(southern 、northern)是一致的,都是应用已知核酸序列作为探针与互补的靶核苷酸序列杂交,通过随后的信号检测进行定性与定量分析,基因芯片在一微小的基片(硅片、玻片、塑料片等)表面集成了大量的分子识别探针,能够在同一时间内平行分析大量的基因,进行大信息量的筛选与检测分析[3,4]。基因芯片主要技术流程包括:芯片的设计与制备;靶基因的标记;芯片杂交与杂交信号检测。 基因芯片的设计实际上是指芯片上核酸探针序列的选择以及排布,设计方法取决于其应用目的,目前的应用范围主要包括基因表达和转录图谱分析及靶序列中单碱基多态位点(single nucleotide polymorphism,SNP)或突变点的检测,表达型芯片的目的是在杂交实验中对多个不同状态样品(不同组织或不同发育阶段、不同药物刺激)中数千基因的表达差异进行定量检测,探针序列一般来自于已知基因的cDNA 或EST库,设计时序列的特异性应放在首要位置,以保证与待测目的基因的特异结合,对于同一目的基因可设计多个序列不相重复的探针,使最终的数据更为可靠。基因单碱基多态检测的芯片一般采用等长移位设计法[5],即按靶序列从头到尾依次取一定长度的互补的核苷酸序列形成一探针组合,这组探针是与靶序列完全匹配的野生型探针,然后对于每一野生型探针,将其中间位置的某一碱基分别用其它三种碱基替换,形成三种不同的单碱基变化的核苷酸探针,这种设计可以对某一段核酸序列所有可能的SNPs位点进行扫描。 芯片制备方法主要包括两种类型:(1)点样法:首先是探针库的制备, 根据基因芯片的分析目标从相关的基因数据库中选取特异的序列进行PCR扩增或直接人工合成寡核苷酸序列[6],然后通过计算机控制的三坐标工作平台用特殊的针头和微喷头分别把不同的探针溶液逐点分配在玻璃、尼龙以及其它固相基片表面的不同位点上,通过物理和化学的方法使之固定,该方法各技术环节均较成熟,且灵活性大,适合于研究单位根据需要自行制备点阵规模适中的基因芯片。(2)原位合成法[7~10]:该法是在玻璃等硬质表面上直接合成寡核苷酸探针阵列,目前应用的主要有光去保护并行合成法,压电打印合成法等,其关键是高空间分辨率的模板定位技术和高合成产率的DNA化学合成技术,适合制作大规模DNA探针芯片,实现高密度芯片的标准化和规模化生产。待分析样品的制备是基因芯片实验流程的一个重要环节, 靶基因在与芯片探针结合杂交之前必需进行分离、扩增及标记。标记方法根据样品来源、芯片类型和研究目的的不同而有所差异。通常是在待测样品的PCR扩增、逆转录或体外转录过程中实现对靶基因的标记。对于检测细胞内mRNA表达水平的芯片,一般需要从细胞和组织中提取RNA,进行逆转录,并加入偶联有标记物的dNTP,从而完成对靶基因的标记过程[11],对于阵列密度较小的芯片可以用同位素,所需仪器均为实验室常规使用设备,易于开展相关工作,但是在信号检测时,一些杂交信号强的点阵容易产生光晕,干扰周围信号的分析。高密度芯片的分析一般采用荧光素标记靶基因,通过适当内参的设置及对荧光信号强度的标化可对细胞内mRNA的表达进行定量检测。近年来运用的多色荧光标记技术可更直观地比较不同来源样品的基因表达差异,即把不同来源的靶基因用不同激发波长的荧光素标记,并使它们同时与基因芯片杂交,通过比较芯片上不同波长荧光的分布图获得不同样品间差异表达基因的图谱[12,13],常用的双色荧光试剂有Cy3- dNTP和Cy5- dNTP。对多态性和突变检测型基因芯片采用多色荧光技术可以大大提高芯片的准确性和检测范围,例如用不同的荧光素分别标记靶序列及单碱基失配的参考序列,使它们同时与芯片杂交,通过不同荧光强弱的比较得出靶序列中碱基失配的信息[14]。 基因芯片与靶基因的杂交过程与一般的分子杂交过程基本相同,杂交反应的条件要根据探针的长度、GC碱基含量及芯片的类型来优化,如用于基因表达检测,杂交的严格性较低,而用于突变检测的芯片的杂交温度高,杂交时间短,条件相对严格。如果是用同位素标记靶基因,其后的信号检测即是放射自显影,若用荧光标记,则需要一套荧光扫描及分析系统,对相应探针阵列上的荧光强度进行分析比较,从而得到待测样品的相应信息。由于基因芯片获取的信息量大,对于基因芯片杂交数据的分析、处理、查询、比较等需要一个标准的数据格式,目前,一个大型的基因芯片的数据库正在构建中,将各实验室获得的基因芯片的结果集中起来,以利于数据的交流及结果的评估与分析。
由安徽医科大学第一、第二附属医院等国内30多家单位共同协作,中国科学家通过对近2万份样本进行分析,发现了白癜风的易感基因。此项研发的成功进行,标志着我国白癜风易感基因研发跻身世界领先行列。 白癜风是一种常见的色素脱失性皮肤病,皮肤黑素细胞被破坏,原因不明。目前我国患病人数已超过1000万。该病好发于颜面等暴露部位,严重影响形象美观,甚至毁损患者容貌,并经常合并炎症性肠病、银屑病、糖尿病、恶性贫血及系统性红斑狼疮等多种自身免疫性疾病,严重危害患者身心健康。 此项研究由安徽医科大学第一、第二附属医院、复旦大学华山医院等国内30多家单位共同协作,历时5年,采用国际最先进的全基因组关联分析方法和生物分析技术进行。通过对近2万份样本进行分析,以强有力的证据指出由遗传因素导致的自身免疫异常是白癜风发病的主要原因,首次在国际上明确白癜风是自身免疫性疾病,并构建了第一个亚洲人群白癜风病例对照的全基因组关联分析数据库,为今后白癜风易感基因的深入研究打下坚实的基础。北京时间6月7日凌晨1点,国际著名学术期刊《自然遗传学》在线发表了该项研究的研究成果。专家认为,此研究成果对于解释白癜风的发病机制具有重大意义,并为疾病预警、临床诊断及新药开发奠定了良好的理论基础。
[align=center][b][font=宋体]利用[/font][font='Times New Roman']MGI[/font][font=宋体]平台对大豆进行全基因组重测序分析[/font][/b][/align][b][font=宋体]摘要[/font][/b][font=宋体][font=宋体]:本研究建立了[/font][font=Times New Roman]MGI[/font][font=宋体]平台全基因重测序的方法。[/font][font=Times New Roman]MGI[/font][font=宋体]平台对大豆的全基因进行重测序结果显示,测序数据质量良好,且与参考基因组比对率较高,符合后续分析要求,对其进行[/font][font=Times New Roman]SNP[/font][font=宋体]和[/font][font=Times New Roman]Indel[/font][font=宋体]的变异检测和注释,此结果说明今后可利用[/font][font=Times New Roman]MGI[/font][font=宋体]平台对其它样品进行全基因重测序分析。[/font][/font][b][font=宋体]关键词[/font][/b][font=宋体][font=宋体]:[/font][font=Times New Roman]MGI[/font][font=宋体]平台;全基因重测序[/font][/font][align=center][font='Times New Roman']Whole genome resequencing analysis of soybeans using the MGI platform[/font][/align][b][font='Times New Roman']Abstract:[/font][font=宋体] [/font][/b][font=宋体][font=Times New Roman]In this study, a method for whole gene resequencing on the MGI platform was established. The results of resequencing the whole genes of soybean by MGI platform showed that the sequencing data was of good quality and had a high comparison rate with the reference genome, which met the requirements of subsequent analysis, and the variation detection and annotation of SNP and Indel were carried out, which indicated that the MGI platform could be used to perform whole gene resequencing analysis on other samples in the future.[/font][/font][b][font='Times New Roman']Keywords:[/font][font=宋体] [/font][/b][font=宋体][font=Times New Roman]MGI platform Whole gene resequencing[/font][/font][font='Times New Roman'] [/font][b][font='Times New Roman']1 [font=宋体]研究背景[/font][/font][/b][font='Times New Roman'][font=宋体]大豆是重要的粮食作物和油料作物,也是人类最主要的植物蛋白来源[/font][/font][font=宋体][font=Times New Roman][1][/font][/font][font=宋体][font=宋体]。我国是野生大豆的发源地,有着极其丰富的大豆种质资源基础,但是育种和产量较其他大豆主产国显得略有不足,究其原因是我国对大豆的研究和发掘力度存在不足,因此,对大豆育成品种的改良势在必行。自[/font][font=Times New Roman]2010[/font][font=宋体]年起,大豆群体水平的重测序也全面开展,在大豆的全基因组变异图谱上也得到了一定的研究进展[/font][/font][font=宋体][font=Times New Roman][2][/font][/font][font=宋体][font=宋体]。本研究利用[/font][font=Times New Roman]MGI[/font][font=宋体]平台对大豆全基因组进行重测序分析,挖掘全基因组水平上的突变。[/font][/font][b][font=宋体][font=Times New Roman]2 [/font][font=宋体]实验仪器[/font][/font][/b][font=宋体]主要实验仪器:[/font][font=宋体][font=Times New Roman]MGISP-960[/font][font=宋体]、[/font][font=Times New Roman]MGIDL-T7[/font][font=宋体]、[/font][font=Times New Roman]DNBSEQ-T7[/font][/font][b][font=宋体][font=Times New Roman]3 [/font][font=宋体]实验结果[/font][/font][font=宋体][font=Times New Roman]3.1 [/font][font=宋体]测序数据质量[/font][/font][/b][font=宋体][font=宋体]根据[/font][font=Times New Roman]MGI[/font][font=宋体]平台的测序特点,使用双端测序的数据,要求[/font][font=Times New Roman]Q30[/font][font=宋体]平均比例在[/font][font=Times New Roman]85%[/font][font=宋体]以上,可以看出大豆重测序数据[/font][font=Times New Roman]Q30[/font][font=宋体]平均比例在[/font][font=Times New Roman]94.72%[/font][font=宋体]以上,说明大豆测序数据质量良好,满足分析要求。[/font][/font][font='Times New Roman'] [/font][font='Times New Roman'] [/font][b][font=黑体][font=黑体]表[/font][font=Times New Roman]1 [/font][font=黑体]测序数据统计表[/font][/font][/b][table][tr][td][align=center][font='Times New Roman']Samples[/font][/align][/td][td][align=center][font='Times New Roman']ID[/font][/align][/td][td][align=center][font='Times New Roman']Clean reads[/font][/align][/td][td][align=center][font='Times New Roman']Clean bases[/font][/align][/td][td][align=center][font='Times New Roman']GC Content[/font][/align][/td][td][align=center][font='Times New Roman']%[/font][font=等线]≥[/font][font='Times New Roman']Q20[/font][/align][/td][td][align=center][font='Times New Roman']%[/font][font=等线]≥[/font][font='Times New Roman']Q30[/font][/align][/td][/tr][tr][td][align=center][font='Times New Roman']P117[/font][/align][/td][td][align=center][font='Times New Roman']P117[/font][/align][/td][td][align=center][font='Times New Roman']169494922[/font][/align][/td][td][align=center][font='Times New Roman']25424238300[/font][/align][/td][td][align=center][font='Times New Roman']36.18%[/font][/align][/td][td][align=center][font='Times New Roman']98.49%[/font][/align][/td][td][align=center][font='Times New Roman']95.27%[/font][/align][/td][/tr][tr][td][align=center][font='Times New Roman']P118[/font][/align][/td][td][align=center][font='Times New Roman']P118[/font][/align][/td][td][align=center][font='Times New Roman']166483906[/font][/align][/td][td][align=center][font='Times New Roman']24972585900[/font][/align][/td][td][align=center][font='Times New Roman']36.47%[/font][/align][/td][td][align=center][font='Times New Roman']98.61%[/font][/align][/td][td][align=center][font='Times New Roman']95.70%[/font][/align][/td][/tr][tr][td][align=center][font='Times New Roman']P119[/font][/align][/td][td][align=center][font='Times New Roman']P119[/font][/align][/td][td][align=center][font='Times New Roman']186127112[/font][/align][/td][td][align=center][font='Times New Roman']27919066800[/font][/align][/td][td][align=center][font='Times New Roman']35.89%[/font][/align][/td][td][align=center][font='Times New Roman']98.57%[/font][/align][/td][td][align=center][font='Times New Roman']95.61%[/font][/align][/td][/tr][tr][td][align=center][font='Times New Roman']P120[/font][/align][/td][td][align=center][font='Times New Roman']P120[/font][/align][/td][td][align=center][font='Times New Roman']192397276[/font][/align][/td][td][align=center][font='Times New Roman']28859591400[/font][/align][/td][td][align=center][font='Times New Roman']36.46%[/font][/align][/td][td][align=center][font='Times New Roman']98.22%[/font][/align][/td][td][align=center][font='Times New Roman']94.72%[/font][/align][/td][/tr][tr][td][align=center][font='Times New Roman']P198[/font][/align][/td][td][align=center][font='Times New Roman']P198[/font][/align][/td][td][align=center][font='Times New Roman']141636468[/font][/align][/td][td][align=center][font='Times New Roman']21245470200[/font][/align][/td][td][align=center][font='Times New Roman']37.11%[/font][/align][/td][td][align=center][font='Times New Roman']98.67%[/font][/align][/td][td][align=center][font='Times New Roman']95.84%[/font][/align][/td][/tr][tr][td][align=center][font='Times New Roman']P199[/font][/align][/td][td][align=center][font='Times New Roman']P199[/font][/align][/td][td][align=center][font='Times New Roman']169468714[/font][/align][/td][td][align=center][font='Times New Roman']25420307100[/font][/align][/td][td][align=center][font='Times New Roman']36.55%[/font][/align][/td][td][align=center][font='Times New Roman']98.60%[/font][/align][/td][td][align=center][font='Times New Roman']95.66%[/font][/align][/td][/tr][tr][td][align=center][font='Times New Roman']P200[/font][/align][/td][td][align=center][font='Times New Roman']P200[/font][/align][/td][td][align=center][font='Times New Roman']155078286[/font][/align][/td][td][align=center][font='Times New Roman']23261742900[/font][/align][/td][td][align=center][font='Times New Roman']37.90%[/font][/align][/td][td][align=center][font='Times New Roman']98.77%[/font][/align][/td][td][align=center][font='Times New Roman']96.14%[/font][/align][/td][/tr][/table][font=Calibri] [/font][font=宋体][font=宋体]样品原始数据碱基质量值可由图[/font][font=Times New Roman]1[/font][font=宋体]看出不存在异常碱基,[/font][font=Times New Roman]6[/font][font=宋体]个大豆碱基测序错误率分布均如图[/font][font=Times New Roman]1[/font][font=宋体]。[/font][/font][align=center][img=,321,]file:///C:/Users/xuxu/AppData/Local/Temp/ksohtml9716/wps1.jpg[/img][font=Calibri] [/font][/align][align=center][b][font=黑体][font=黑体]图[/font] [font=Times New Roman]1 [/font][font=黑体]碱基测序错误率分布图[/font][/font][/b][/align][font=宋体][font=宋体]碱基类型分布检查可用于检测有无[/font][font=Times New Roman]AT[/font][font=宋体]、[/font][font=Times New Roman]GC[/font][font=宋体]分离现象,若有碱基分离现象可能是测序或建库所带来的,并会影响后续分析。高通量所测序为基因组随即打断后的[/font][font=Times New Roman]DNA[/font][font=宋体]片段,由于位点在基因组上的分布是近似均匀的,同时,[/font][font=Times New Roman]G/C[/font][font=宋体]、[/font][font=Times New Roman]A/T[/font][font=宋体]含量也是近似均匀的。因此,根据大数定理,在每个测序循环上,[/font][font=Times New Roman]GC[/font][font=宋体]、[/font][font=Times New Roman]AT[/font][font=宋体]含量应当分别相等,且等于基因组的[/font][font=Times New Roman]GC[/font][font=宋体]、[/font][font=Times New Roman]AT[/font][font=宋体]含量。同样因为重叠等的关系会导致样品前几个碱基[/font][font=Times New Roman]AT[/font][font=宋体]、[/font][font=Times New Roman]GC[/font][font=宋体]不等波动较大,高于其他测序区段,而其它区段的[/font][font=Times New Roman]GC[/font][font=宋体]、[/font][font=Times New Roman]AT[/font][font=宋体]含量相等,且分布均匀无分离现象,如图[/font][font=Times New Roman]2[/font][font=宋体]所示。[/font][/font][align=center][img=,321,]file:///C:/Users/xuxu/AppData/Local/Temp/ksohtml9716/wps2.jpg[/img][font=Calibri] [/font][/align][b][font=黑体][font=黑体]图[/font][font=Times New Roman]2 ATGC[/font][font=黑体]含量分布图[/font][/font][font=宋体][font=Times New Roman]3.2 [/font][font=宋体]与参考基因组的序列比对[/font][/font][font='Times New Roman']3.2.1 [font=宋体]比对结果[/font][/font][/b][font=宋体][font=宋体]将测序得到的大豆样品与参考基因进行序列比对,[/font][font=Times New Roman]bwa[/font][font=宋体]软件主要用于二代高通量测序得到的短序列与参考基因组进行比对,比对结果见表[/font][font=Times New Roman]2[/font][font=宋体],根据比对结果可评估测序数据是否满足后续分析。[/font][/font][align=center][b][font=黑体][font=黑体]表[/font][font=Times New Roman]2 [/font][font=黑体]比对效率统计表[/font][/font][/b][/align][table][tr][td][align=center][font='Times New Roman']Sample_ID[/font][/align][/td][td][align=center][font='Times New Roman']Mapped(%)[/font][/align][/td][td][align=center][font='Times New Roman']Properly_mapped(%)[/font][/align][/td][td][align=center][font='Times New Roman']Averge_depth[/font][/align][/td][/tr][tr][td][align=center][font='Times New Roman']P117[/font][/align][/td][td][align=center][font='Times New Roman']99.99%[/font][/align][/td][td][align=center][font='Times New Roman']98.53%[/font][/align][/td][td][align=center][font='Times New Roman']25.44[/font][/align][/td][/tr][tr][td][align=center][font='Times New Roman']P118[/font][/align][/td][td][align=center][font='Times New Roman']99.99%[/font][/align][/td][td][align=center][font='Times New Roman']98.55%[/font][/align][/td][td][align=center][font='Times New Roman']24.9[/font][/align][/td][/tr][tr][td][align=center][font='Times New Roman']P119[/font][/align][/td][td][align=center][font='Times New Roman']99.99%[/font][/align][/td][td][align=center][font='Times New Roman']98.63%[/font][/align][/td][td][align=center][font='Times New Roman']27.75[/font][/align][/td][/tr][tr][td][align=center][font='Times New Roman']P120[/font][/align][/td][td][align=center][font='Times New Roman']99.98%[/font][/align][/td][td][align=center][font='Times New Roman']98.28%[/font][/align][/td][td][align=center][font='Times New Roman']28.58[/font][/align][/td][/tr][tr][td][align=center][font='Times New Roman']P198[/font][/align][/td][td][align=center][font='Times New Roman']99.99%[/font][/align][/td][td][align=center][font='Times New Roman']98.58%[/font][/align][/td][td][align=center][font='Times New Roman']21.26[/font][/align][/td][/tr][tr][td][align=center][font='Times New Roman']P199[/font][/align][/td][td][align=center][font='Times New Roman']99.98%[/font][/align][/td][td][align=center][font='Times New Roman']98.50%[/font][/align][/td][td][align=center][font='Times New Roman']25[/font][/align][/td][/tr][tr][td][align=center][font='Times New Roman']P200[/font][/align][/td][td][align=center][font='Times New Roman']99.99%[/font][/align][/td][td][align=center][font='Times New Roman']98.13%[/font][/align][/td][td][align=center][font='Times New Roman']23.13[/font][/align][/td][/tr][/table][font=宋体][font=宋体]将比对到不同染色体的[/font][font=Times New Roman]Reads[/font][font=宋体]进行位置分布统计,绘制[/font][font=Times New Roman]Mapped Reads[/font][font=宋体]在参考基因组上的覆盖深度分布图,见图[/font][font=Times New Roman]3[/font][font=宋体]。[/font][/font][align=center][img=,321,]file:///C:/Users/xuxu/AppData/Local/Temp/ksohtml9716/wps3.jpg[/img][font=Calibri] [/font][/align][align=center][b][font=黑体][font=黑体]图[/font][font=Times New Roman]3 Mapped Reads[/font][font=黑体]在参考基因组上的位置及覆盖深度分布图[/font][/font][/b][/align][font=宋体][font=宋体]统计[/font][font=Times New Roman]Mapped Reads[/font][font=宋体]在指定的参考基因组不同区域的数目,绘制基因组不同区域样品[/font][font=Times New Roman]Mapped Reads[/font][font=宋体]的分布图,见图[/font][font=Times New Roman]4[/font][/font][align=center][img=,321,]file:///C:/Users/xuxu/AppData/Local/Temp/ksohtml9716/wps4.jpg[/img][font=Calibri] [/font][/align][b][font=黑体][font=黑体]图[/font][font=Times New Roman]4 [/font][font=黑体]基因组不同区域[/font][font=Times New Roman]Reads[/font][font=黑体]分布图[/font][/font][font=宋体][font=Times New Roman]3.2.2 [/font][font=宋体]插入片段长度检验[/font][/font][/b][font=宋体][font=宋体]通过检测双端序列在参考基因组上的起止位置,可以得到样品[/font][font=Times New Roman]DNA[/font][font=宋体]打断后得到的测序片段的实际大小,即插入片段大小([/font][font=Times New Roman]Insert Size[/font][font=宋体]),它是信息分析时的一个重要参数。插入片段大小的分布一般符合正态分布,且只有一个单峰,[/font][font=Times New Roman]Insert Size[/font][font=宋体]分布图可以展示各个样品的插入片段的长度分布情况。各样品的插入片段长度模拟分布图见图[/font][font=Times New Roman]5[/font][font=宋体]。[/font][/font][align=center][img=,321,]file:///C:/Users/xuxu/AppData/Local/Temp/ksohtml9716/wps5.jpg[/img][font=Calibri] [/font][/align][align=center][b][font=黑体][font=黑体]图[/font][font=Times New Roman]5 [/font][font=黑体]插入片段长度模拟图[/font][/font][/b][/align][b][font=宋体][font=Times New Roman]3.2.3[/font][/font][font='Times New Roman'][font=宋体]深度分布统计图[/font][/font][/b][font='Times New Roman']Reads[font=宋体]定位到参考基因组后,可以统计参考基因组上碱基的覆盖情况。参考基因组上被[/font][font=Times New Roman]reads[/font][font=宋体]覆盖到的碱基数占基因组的百分比称为基因组覆盖度;碱基上覆盖的[/font][font=Times New Roman]reads[/font][font=宋体]数为覆盖深度。基因组覆盖度可以反映参考基因组上变异检测的完整性,覆盖到的区域越多,可以检测到的变异位点也越多。[/font][/font][font='Times New Roman'][font=宋体]覆盖度主要受测序深度以及样品与参考基因组亲缘关系远近的影响。基因组的覆盖深度会影响变异检测的准确性,在覆盖深度较高的区域(非重复序列区),变异检测的准确性也越高。[/font][/font][font='Times New Roman'][font=宋体]另外,若基因组上碱基的覆盖深度分布较均匀,也说明测序随机性较好。样品的碱基覆盖深度分布曲线和覆盖度分布曲线见图[/font][/font][font=宋体][font=Times New Roman]6[/font][font=宋体]。[/font][/font][align=center][img=,321,]file:///C:/Users/xuxu/AppData/Local/Temp/ksohtml9716/wps6.jpg[/img][font=Calibri] [/font][/align][align=center][b][font=黑体][font=黑体]图[/font] [font=Times New Roman]6 [/font][font=黑体]深度分布统计图[/font][/font][/b][/align][b][font=宋体][font=Times New Roman]3.3 [/font][font=宋体]变异检测[/font][/font][font=宋体][font=Times New Roman]3.3.1 SNP[/font][font=宋体]检测与注释[/font][/font][/b][font='Times New Roman'][font=宋体]根据变异位点在参考基因组上的位置以及参考基因组上的基因位置信息,可以得到变异位点在基因组发生的区域(基因间区、基因区或[/font]CDS[font=宋体]区等),以及变异产生的影响(同义非同义突变等)。软件可以使用[/font][font=Times New Roman]vcf[/font][font=宋体]格式文件作为输入和输[/font][/font][font=宋体][font=宋体]出,见图[/font][font=Times New Roman]7[/font][font=宋体]和图[/font][font=Times New Roman]8[/font][font=宋体]。[/font][/font][align=center][img=,321,]file:///C:/Users/xuxu/AppData/Local/Temp/ksohtml9716/wps7.jpg[/img][font=Calibri] [/font][/align][align=center][b][font=黑体][font=黑体]图[/font][font=Times New Roman]7 SNP[/font][font=黑体]突变类型分布图[/font][/font][/b][/align][align=center][img=,344,]file:///C:/Users/xuxu/AppData/Local/Temp/ksohtml9716/wps8.jpg[/img][font=Calibri] [/font][/align][b][font=黑体][font=黑体]图[/font][font=Times New Roman]8 SNP[/font][font=黑体]注释分类图[/font][/font][font=宋体][font=Times New Roman]3.3.2 Indel[/font][font=宋体]检测与注释[/font][/font][/b][font=宋体][font=宋体]根据所有样品在[/font][font=Times New Roman]CDS[/font][font=宋体]区和全基因范围的[/font][font=Times New Roman]Indel[/font][font=宋体]长度进行统计,其长度分布如图[/font][font=Times New Roman]9[/font][font=宋体]。[/font][/font][align=center][img=,355,]file:///C:/Users/xuxu/AppData/Local/Temp/ksohtml9716/wps9.jpg[/img][font=Calibri] [/font][/align][align=center][b][font=黑体][font=黑体]图[/font][font=Times New Roman]9 [/font][font=黑体]全基因和编码区[/font][font=Times New Roman]Indel[/font][font=黑体]长度分布图[/font][/font][/b][/align][font='Times New Roman'][font=宋体]根据样品检测得到的[/font]Ind[/font][font=宋体][font=Times New Roman]el[/font][/font][font='Times New Roman'][font=宋体]位点在参考基因组上的位置信息,对比参考基因组的基因、[/font]CDS[font=宋体]位置等信息,可以注释[/font][font=Times New Roman]Indel[/font][font=宋体]位点是否发生在基因间区、基因区或[/font][font=Times New Roman]CDS[/font][font=宋体]区、是否为移码突变等。发生移码突变的[/font][font=Times New Roman]Indel[/font][font=宋体]可能会导致基因功能的改变,具体注释结果见[/font][/font][font=宋体][font=宋体]图[/font][font=Times New Roman]10[/font][font=宋体]。[/font][/font][align=center][img=,344,]file:///C:/Users/xuxu/AppData/Local/Temp/ksohtml9716/wps10.jpg[/img][font=Calibri] [/font][/align][align=center][b][font=黑体][font=黑体]图[/font] [font=Times New Roman]10 Indel [/font][font=黑体]注释分类图[/font][/font][/b][/align][b][font=宋体][font=Times New Roman]4 [/font][font=宋体]结论[/font][/font][/b][font=宋体][font=宋体]本文基于[/font][font=Times New Roman]MGI[/font][font=宋体]对大豆进行重基因测序,实验结果可看出,大豆样品测序产出数据良好,与参考基因组序列比对率较高,符合后续分析,对其进行变异检测可得到[/font][font=Times New Roman]SNP[/font][font=宋体]和[/font][font=Times New Roman]Indel[/font][font=宋体]的结果。其它研究表明[/font][/font][font=宋体][font=Times New Roman]MGISEQ-2000[/font][font=宋体]全基因组重测序表现性能稳定、质量可靠,在实际应用上有明显的优势和应用价值[/font][font=Times New Roman][3][/font][font=宋体]。对[/font][/font][font=宋体][font=宋体]本次实验说明[/font][font=Times New Roman]MGI[/font][font=宋体]平台对样品进行重测序效果良好,后续可对其它植物进行重测序。[/font][/font][font=宋体] [/font][font=宋体] [/font][font=宋体]参考文献:[/font][font=宋体][font=Calibri][1] [/font][/font][font='Times New Roman'][font=宋体]张永芳[/font],[font=宋体]钱肖娜[/font][font=Times New Roman],[/font][font=宋体]王润梅[/font][/font][font=宋体][font=Times New Roman],[/font][font=宋体]等[/font][font=Times New Roman]. [/font][font=宋体]不同大豆材料的抗旱性鉴定及耐旱品种筛选[/font][font=Times New Roman][J].[/font][font=宋体]作物杂志[/font][font=Times New Roman],2019(5): 41-45.[/font][/font][font=宋体][font=Calibri][2] [/font][font=宋体]邬启帆[/font][font=Calibri]. [/font][font=宋体]基于基因组重测序黄淮海大豆育成品种遗传结构及重要家族遗传基础研究[/font][font=Calibri][D]. [/font][font=宋体]南昌[/font][/font][font=宋体][font=宋体]大学[/font][font=Times New Roman], 2023.[/font][/font][font=宋体][font=Calibri][3] [/font][/font][font=宋体][font=宋体]李伟宁[/font][font=Times New Roman],[/font][font=宋体]刘刚[/font][font=Times New Roman],[/font][font=宋体]周荣等[/font][font=Times New Roman]. MGISEQ-2000[/font][font=宋体]、[/font][font=Times New Roman]HiSeq 2000[/font][font=宋体]与[/font][font=Times New Roman]NovaSeq 6000[/font][font=宋体]平台全基因组重测序数据的比较分析[/font][font=Times New Roman][J]. [/font][font=宋体]中国畜牧杂志[/font][font=Times New Roman],2021,57(11):156-162.[/font][/font]
科学家发现一种基因识别新技术科学家们发现了一项基因识别新技术,能将我们掌握的动物遗传信息增加70-80%。研究结果发表在《自然—方法学》(Nature Methods)杂志上,有可能彻底改变我们对于动物遗传学和疾病的认识,并提高我们对于如SARS等跨越物种障碍由动物向人类传播的危险病毒的认识。现代基因组测序技术的进步使得科学家们能够揭示各种各样动物、植物和昆虫的遗传密码,确定控制一切事物,从我们的眼睛颜色到对某些疾病易感性的遗传信息和变异。直到现在,正确识别隐藏在新测序物种遗传物质中的基因和蛋白质还是一项艰巨的任务,需要细致的观察以及编撰大量构成任一动物、植物或昆虫的成千上万基因的数据。论文的主要作者、布里斯托大学细胞与分子医学院的高级讲师David Matthews说:“基因识别主要是借助计算机程序搜寻与在其他动物或人类中已经发现的基因相似的基因组区域。然而,这种分析并不总是有效。”布里斯托大学研究小组现在发现了一种更有效的方法:测序表达的mRNA生成蛋白质数据库再进行质谱分析。这种将高通量测序与蛋白质鉴别技术相结合的方法可以直接观察生成的基因和所有蛋白质,检测存在于动物、植物和昆虫中的遗传信息。为了证实他们的技术起作用,研究人员进行了一项实验,验证他们的程序在基因发现方面的能力。他们用一种充分了解的感冒病毒模拟新发现的病毒感染人类细胞。随后采用新技术分析了这些感染细胞。 当与人类和感冒病毒的已知遗传信息进行比较时,由此生成的“发现”基因和蛋白质的列表证实是极其成功的,并且证明了这一方法的效力。对于仓鼠细胞的类似分析提供了直接观察的证据,在一项相对廉价的实验中研究人员证实仓鼠存在数以千计的基因和蛋白。这些仓鼠中几乎所有基因和蛋白质的直接证据均无法在仓鼠基因和蛋白质的“官方”列表上获得。Matthews博士补充说:“这些研究发现为利用当前强有力的分析工具研究人类疾病,并将它们应用于动物、昆虫或甚至植物——研究一些以往非常具有挑战性或根本不可能的事物开辟了可能性。这一技术也将使得科学家们能够更容易更有效的研究从农场动物及其疾病到危害农作物的病虫害等一切事物。”近年来,包括流感、SARS、埃博拉病毒(Ebola virus)、亨德拉病毒(Hendra virus)和尼帕病毒(Nipah virus)等许多危险的新病毒从动物向人类传播。今年早些时候在中东有三人接触了一种被认为是直接来自蝙蝠的新SARS样病毒而患重病,其中两人死亡。“由于对这些生物体的遗传构成所知甚少,为何这些病毒对蝙蝠的疾病影响有限是一个待解谜题。我们开始将我们的技术应用于实验室培养的蝙蝠细胞,通过分析蝙蝠的遗传和蛋白质含量更多地认识它们的遗传学,了解它们是如何能够与常常对人类造成致命后果的这些病毒明显共存的。
科技日报讯 (记者王怡)中国科学院北京基因组研究所和吉林中科紫鑫科技有限公司4月18日在长春联合召开国产新一代基因测序仪样机观摩研讨会,向与会的国内基因测序领域专家和应用单位代表展示这一合作成果,同时向社会公开征集部分应用单位进行免费测试使用,测试工作将于今年下半年开展。 据了解,新一代基因测序仪样机是目前唯一技术性可以匹敌国际市场主流产品的国产基因测序系统,与国外第二代高通量测序系统相比,已经成功解决“读长较短”这一关键技术难题。该基因测序仪已经达到和部分超越国际主流设备技术指标,其成本低于进口设备1/3以上,应用成本低于进口设备1/5以上,使新一代测序技术真正达到进入广泛应用市场的经济条件,并将彻底解决我国基因测序仪完全依赖进口的局面。截至目前,该系统已获得7个发明专利和1个实用新型专利授权,还有多项专利正在申报。 中科院北京基因组研究所研究员于军介绍,新一代基因测序仪对传染性疾病的预防控制和诊疗,生物恐怖因子、食源性致病因子和转基因成分鉴定,口岸卫生和有害生物防御性检疫,以及针对人类遗传多样性而产生的疾病早期预警和个体化用药相关基因的检测分析等实践应用,提供强有力的技术支持。 据悉,测序仪今年下半年将在医疗、检验检疫、疾病防控、高校、研究院所等20家应用单位进行免费测试使用。已经确认参加测试应用的单位包括中国检验检疫科学研究院、北京出入境检验检疫局、青岛海洋大学等10余家单位。测试工作主要包括对系统性能的优化和改进,以及根据应用领域的不同进行应用产品的共同开发,即在基础试剂产品的平台上衍生出一系列专用型试剂产品,并开发相应的数据分析算法和数据库,实现测序技术实践应用的全面解决方案。来源:中国科技网-科技日报 2014年04月20日
第三张“基因变异图谱”与第二代基因组测序技术——评“千人基因组计划”首期研究成果的医学意义世界上任意两个人的基因99%都是相同的,而恰是那1%不同,负责着个体间的表型差异。《自然》杂志近期披露,当人体内携带有250到300基因变异位点的时候,相关基因就就会“沉默”。甚至,一个人只携带了 50到100基因变异位点,就可能患上某种疾病。10年前,“人类基因组计划”这一耗资30亿美元、历时10余年的伟大科学工程完成之际,人们以为得到了揭开自身生命奥秘的天书,生命科学也划时代地进入了“后基因组时代”。如今看来,当时得到的仅仅是人类基因组的“参考图谱”,对于人群里个体间的基因差异,或是更具医学意义的“基因变异图谱”来说,人们知之甚少。第三张“基因变异图谱”为了探寻个体间的基因差异,科学界在2002年启动了HapMap(人类基因组单体型图谱)计划。Hapmap在2005年完成的“第一张基因变异图谱”含有一百万个“单核苷酸多态性”(SNPs)位点;HapMap在2008年完成的“第二张基因变异图谱”含有三百一十万个SNPs位点。而此次“千人基因组”所公布的一期结果——“第三张基因变异图谱”,已经包含了一千五百万个SNPs位点。今年10月28日,《自然》杂志为此刊出的文章题目为“基于群体规模的基因变异图谱”,鲜明的指出,“千人基因组计划”首期研究成果,其最大优势在于:“第三张基因变异图谱”所采用的样本,针对了“大规模人群”。 远超过此前两张“基因变异图谱”所测定的样本数。绘制“第三张基因变异图谱”的所有数据,是基于两个核心家庭,6个个体的精确基因组测序,179个个体的低覆盖率基因组测序,以及七百多人的蛋白编码区的基因测序。检测人群数目庞大,人种涉及中国人、日本人、西欧人等。因此,第三张“人类基因变异图谱”的问世,可以从更深的层次上了解,种族之间、个体之间的基因差异。更具医学意义的是,对于人群中发生频率在1%以上的基因变异,本次研究的覆盖率达到95%以上。这就意味着:此前Hapmap计划所绘制的两张“基因变异图谱”中,没能涉及的“罕见病”致病基因,可能在“第三张基因变异图谱”中已经被标出。“基因变异图谱”的医学应用随着,“人类基因变异图谱”绘制的日臻完善,和商业化全基因组SNP 分型芯片成本的不断降低,以及新的统计方法和软件的出现, “全基因组关联分析”( Genome-Wide Associat ion Study , GWAS) 越来越多的应用于复杂疾病“易感基因”的确定。今年6月6日,安徽医科大学的张学军教授领衔的团队,通过对中国汉族和维吾尔族人群近2万份样本进行分析,在人类基因组的3个区域内发现与白癜风发病密切相关的4个易感基因。今年8月2日,中***事医学院贺福初院士领衔的蛋白质组学国家重点实验室,通过对大陆5个肝癌高发区的4500多名肝癌病例和对照的研究,发现了肝癌易感基因新区域(1p36.22)今年8月23日,新乡医学院的王立东教授联合国内18家医院,建立了数十万份的食管癌标本资料库,并首次在人类第10号和20号染色体上,发现两个食管癌易感基因(PLCE1和C20orf54)。基因变异有着很强的人种差异,相比国外此领域的研究成果,以上研究成果的临床意义,在于其是针对我国的特有人群。也就是说,以上研究成果在我国的临床上更具医学价值。更为可喜的是,以上研究成果均发表在此领域最为权威的《自然 遗传学》杂志上。我国在利用GWAS需找复杂疾病易感基因领域的研究,已经得到了世界的公认。
转基因食品用什么仪器和分析试剂能检测出来?
最新发现与创新 中国科技网讯 日前,解放军307医院免疫室及国家生物医学分析中心免疫室主任、著名血液免疫学家奚永志教授申报的《编码鸡Ⅱ型胶原CCOL2A1全长基因及其用途》基因发明,获得欧洲基因发明专利。这是我国迄今为止获得的国际公认具有重大药用价值的首个欧洲基因发明专利。 奚永志已于近日收到欧洲专利局的正式公文通知:“根据欧洲专利法第123(2)款之规定,奚永志教授申报的《编码鸡Ⅱ型胶原CCOL2A1全长基因及其用途》基因发明申请具有新颖性、创造性和工业实用性,该发明攻克了国际上该领域长期难以解决的重大关键难题。权利要求1—8是可授权允许的。”能够在基因科学研究领域获得我国具有完全自主知识产权的欧洲基因发明专利,标志着我国在基因科学研究领域的突破,彰显了我国在基因科学研究领域原始创新能力的提升,打破了欧美发达国家在基因发明专利上的长期垄断。 据悉,奚永志获得这项欧洲基因发明专利是拥有国际标准的“三方专利”优势。“三方专利”被国际上列为评定创新型国家不可或缺的四个重要标志之一。这项基因发明专利,可用于研制开发有效治疗类风湿关节炎(RA)的基因药物和基因治疗。在科技部“‘十一五’重大新药创制”科技重大专项的资助下,该课题组采用这个稀有“珍贵基因”又率先原创性地研制成功国际上首个基于异种CCOL2A1能有效治疗RA的全新型治疗性基因疫苗,获得中国发明专利。目前该课题组正在积极完善新药临床前相关研究和进行临床试验报批工作,力争早日用于临床,造福社会。(记者 张克 通讯员 张国清) 《科技日报》(2012-09-21 一版)
不久前,俄罗斯宣布取消对耐草甘膦转基因玉米NK603的临时禁令。至此,去年下半年出现的关于转基因玉米有毒甚至致癌的传言又算画上了句号。 事实上,自从转基因作物引入种植以来,关于它的各种流言就层出不穷。在日前中国科协举办的《科学家与媒体面对面·转基因技术安全管理》活动上,有关专家对此进行了分析和解读。 流言:转基因玉米容易致癌? 真相:实验设计有严重漏洞! 最新的一起关于转基因作物的流言,来自于去年9月19日法国卡昂大学分子生物学家塞拉利尼等人在英国期刊发表的一份研究报告。报告称,其长达两年的研究显示,喂食美国孟山都公司NK603转基因玉米的实验鼠寿命比正常实验鼠短,且前者出现肿瘤的几率更高。该报告对已经在欧盟获准上市的这种转基因玉米的安全性提出质疑。 事件发生后,俄罗斯相关部门反应十分谨慎,决定暂停进口和使用转基因玉米品种NK603. 法国国家卫生安全署、生物技术最高委员会和欧洲食品安全局均对塞拉利尼等人的研究展开调查。去年11月,欧洲食品安全局作出最终评估,彻底否定了这种转基因玉米有毒甚至致癌的研究结论。 欧洲食品安全局认为,卡昂大学研究人员得出的研究结论不仅缺乏数据支持,而且相关实验的设计和方法都存在严重漏洞,这些问题说明,可接受的科研标准在实验中没有得到遵守。该局要求研究负责人提供更多相关信息,以增强报告的可信度。但这一要求被塞拉利尼拒绝。 有人指出,实验所用的老鼠类型本身易患癌。 俄罗斯有关部门也对此进行了安全性评估,结论认为,转基因玉米NK603与其常规品种中的化学组分等同;其中的转基因蛋白既不对人体有毒,也不是过敏源;未发现其具有任何毒性、遗传毒性、致敏性、过敏和免疫调节作用;目前,转基因玉米NK603经17个国家核准登记,并在饮食中获准使用,未发现其对人体健康有不良影响。基于此,俄罗斯已于日前取消了对转基因玉米NK603的临时禁令。 流言:转基因大豆引发过敏? 真相:实验室阶段就已中止! 其实,早在1994年,关于转基因作物的流言就已经出现了。 大豆是富含氨基酸的营养食物。但在大豆的氨基酸中缺乏含硫氨基酸。而巴西坚果中有一种富含甲硫氨基酸的蛋白。为了进一步提高大豆的营养品质,1994年,美国先锋种子公司的科研人员就尝试将巴西坚果中的这种蛋白转入大豆中。但进一步的实验表明,对巴西坚果过敏的人同样对这种大豆过敏,而转入的这种蛋白质可能正是主要过敏源。基于此,先锋种子公司立即停止这项研究计划。 然而,这件事后来被一些人说成"转基因大豆可以引起食物过敏",成为反对转基因的一个主要事例。中国农科院生物技术研究所陈茹梅研究员指出,转基因技术本身是中性的,可以用它来做好事,也可以用它来做坏事。正因为如此,各个国家对于转基因作物的安全管理都有严格的要求。而"巴西坚果事件",恰恰是转基因技术管理的成功案例。 流言:转基因马铃薯造成消瘦? 真相:单吃淀粉老鼠也受不了! 1998年秋天,苏格兰罗威特研究所的普斯泰博士通过电视台发表讲话,声称在实验中用转雪花莲凝集素基因的马铃薯喂食大鼠,大鼠"体重和器官重量严重减轻,免疫系统受到破坏".此言一出,立刻引发了反对转基因技术的一轮热潮。 英国皇家学会对"普斯泰事件"高度重视,组织专家对该实验展开同行评审。1999年5月,评审报告指出普斯泰的实验存在失误和缺陷,主要包含六个方面: 不能确定转基因与非转基因马铃薯的化学成分有差异; 对实验用的大鼠仅仅食用富含淀粉的转基因马铃薯,未补充其它蛋白质以防止饥饿是不适当的; 供实验用的大鼠数量太少,且使用食物都不是大鼠的标准食物,欠缺统计学意义; 实验设计差,未进行双盲测定; 统计方法不恰当; 实验结果无一致性。 不久后,普斯泰博士为自己不负责任的说法表示道歉。罗威特研究所宣布普斯泰提前退休,并不再对其言论负责。 流言:抗虫转基因玉米危害帝王蝶? 真相:野外帝王蝶并不吃玉米花粉! 帝王蝶是美国民众十分喜爱的一种野外观赏昆虫。1999年,美国康奈尔大学昆虫学教授洛希发表文章称帝王蝶在对抗害虫的同时,也对非目标昆虫产生威胁。在实验室中用拌有转基因抗虫玉米花粉的饲料喂帝王蝶幼虫,死亡率高达44%. 转基因抗虫玉米本来的培育目的就是为了对抗害虫,帝王蝶作为一种昆虫,吃多了这种玉米花粉会死,其实并不奇怪。问题是,转基因抗虫玉米真的会对帝王蝶产生巨大威胁吗? 美国环境保护局组织昆虫专家对帝王蝶问题展开专题研究,得出的主要结论是,野外帝王蝶通常不吃玉米花粉,因为它们在玉米散粉之后才大量产卵。而事实上,在所调查的美国中西部田间,转基因抗虫玉米地占总玉米地面积的25%,而田间帝王蝶的数量很大,并未受到影响。 流言:转基因玉米致精液异常? 真相:纯属张冠李戴子虚乌有! 2010年2月起,一篇题为《广西抽检男生一半精液异常,传言早已种植转基因玉米》的帖子在网络上流传甚广,引起了不少公众对转基因产品的恐慌。 2010年3月3日,农业部农业转基因生物安全管理办公室负责人在接受采访时表示,农业部从未批准任何一种转基因粮食种子进口到中国境内进行种植,在国内也没有转基因粮食作物种植。 而广西抽检男生一半精液异常的说法则确有出处,来自广西医科大学第一附属医院男性学科主任梁季鸿等人完成的《广西在校大学生性健康调查报告》,而研究者根本没有提出广西大学生精液异常与转基因有关的观点,而是列出了环境污染、食品中大量使用添加剂、长时间上网等不良习惯的因素。 专家观点 为什么公众 对转基因如此敏感? 中国农业科学院副院长、工程院院士吴孔明指出,我国作为发展中的大国,面临的资源约束是非常明显的。只有依靠技术创新才能支撑我们的粮食、食品需求。而转基因技术是农业科技发展的必由之路,我们没有太多时间去等待。 事实上,我国在推广转基因作物生产方面是十分谨慎的,从某种意义上说已经落后了。比如按照2010年的数据,我们国家在转基因作物种植的面积上,只排在世界第六位,这和我们国家的人口与经济规模是不相称的。历史的经验证明,落后就要挨打,这从我国的大豆生产受到美国转基因大豆的全面排挤中,就可以看得很清楚。 那么,为什么有些公众对转基因食品格外敏感,甚至有着本能的不信任呢? 农业部科技发展中心副主任周云龙对此做了分析和总结,提出了以下几点原因: 转基因技术涉及生物本身甚至人本身的改变,容易引起心理上的抵触; 公众对看不见摸不着的东西有着本能的怀疑和回避; 转基因作物一旦推广,会涉及到每一个人,而且往往只能被动接受,难以主动选择; 转基因技术确实可以用来做坏事,无论是有心还是无意,风险的确存在。如果没有严格的控制和管理,必然会带来食品安全与环境污染方面的问题; 有些人出于狭隘的心理,认为转基因技术是少数大国、大垄断公司的专利。但事实上,技术发展属于全人类。我国在转基因技术发展上也已进入了世界先进水平。 周云龙介绍说,我们国家在转基因技术、产品管理上,无论是理念还是方法,都是和世界发达国家接轨的。而且在某些方面,我们的管理标准更严格。(转自:中国技术性贸易措施网)
八、基因芯片的应用(一)基因表达分析基因芯片具有高度的敏感性和特异性,它可以监测细胞中几个至几千个mRNA拷贝的转录情况。与用单探针分析mRNA的点杂交技术不同,基因芯片表达探针阵列应用了大约20对寡核苷酸探针来监测每一个mRNA的转录情况。每对探针中,包含一个与所要监测的mRNA完全吻合和一个不完全吻合的探针,这两个探针的差别在于其中间位置的核苷酸不同。这种成对的探针可以将非特异性杂交和背景讯号减小到最低的水平,由此我们就可以确定那些低强度的mRNA。目前,Affymetrix公司已经生产出HugeneFL、Mu6500(含有小鼠6500个基因)、Ye6100(含有酵母6100个基因)等基因芯片成品。1.分析基因表达时空特征。英国剑桥大学Whitehead研究所的Frank C.P. Holstege等人,应用含有酵母基因组的基因芯片,深入研究了真核细胞基因组的调节周期。应用基因组水平的表达分析,监测那些表达受转录起始机制的关键成分控制的基因,发现RNA聚合酶II、主要的转录因子TFIID和SAGA染色体修饰复合物等均在基因的表达中有自己特定的作用位点。通过本试验,研究人员揭示了:(1)基因特异性的转录因子对表达的调控作用。(2)细胞在缺乏营养的环境中,基因不同位点的协同调节作用的全新机制。(3)信号转导通路的最终作用位点,在最初的几步中就可以确定。以此试验为基础,研究人员进一步绘制出了酵母基因组控制图,并由此分析出了各种调节因子在基因上不同的作用位点和其作用的分子机制。美国Stanford大学的V.R.Iyer等人,对成纤维细胞中与细胞增生和损伤修复有关的基因进行了分析。首先,他们用成纤维细胞中的8600个基因片断制成基因芯片的探针阵列,通过与mRNA反转录形成的cDNA的杂交反应,可以判断出该基因的活性。在试验中,成纤维细胞被置于无营养的环境中,使绝大部分基因的活性关闭,两天后,加入10%的血清,24小时内,分6个不同的时间点,观察基因的活化情况。试验结果表明,在所有被监测的基因中,约有500个基因最为活跃,而使细胞保持不分裂状态的基因活性被抑制。其中,最早被活化的是那些转录调控基因。在活化的基因中,有28个基因共同作用,控制细胞的增殖;8个与免疫反应的激活有关;19个与血管重建有关;另有许多基因,与血管新生密切相关。在肿瘤细胞中,基因的表达与正常的细胞存在着明显的差异。通过基因芯片绘出基因表达的时空图谱,有助于人类认识生命活动过程和特征。2.基因差异表达检测生命活动中基因表达的改变是生物学研究的核心问题。理解人类基因组中10万个不同的基因功能,监测某些组织、细胞不同分化阶段的差异基因表达(differential gene expression ,DGE)十分重要。对差异表达的研究,可以推断基因与基因的相互关系,细胞分化中基因“开启”或“关闭”的机制;揭示基因与疾病的发生、发展、转归的内在联系。目前DGE研究方法主要有表达序列标签(ESTs)测序、差减克隆(subtractive cloning )、差异显示(differential display)、基因表达系列分析 (serial analysis of gene expression,SAGE)。而cDNA微阵列杂交技术可监测大量mRNA的转录,直接快速地检测出极其微量的mRNA,且易于同时监测成千上万的基因,是研究基因功能的重要手段之一。Rihn BH等利用基因芯片检测胸膜间皮瘤与正常细胞间比较了6500个基因,,发现了300多个差异基因的表达。其中几个典型基因的表达经RT-PCR进行定量后,可作为胸膜间皮瘤诊断的标记物(Markers)。Sgroi报告DNA芯片结合激光捕获显微切割技术(laser capture microdissection)用于乳癌浸润期和转移期及正常细胞的基因表达谱(gene expression profiles)差异研究,结果被定量PCR和免疫组化所证实。差异表达有助于早期发现瘤细胞3万个基因与正常细胞的区别,有助于了解瘤细胞的发生、浸润、转移和药敏。最近,美国毒物化学研究所(CIIT) 和国家环境健康科学研究所(NIEHS)正计划在一张玻片上建立8700个小白鼠cDNA芯片,用于肝癌的研究。我国也已成功研制出能检出41000种基因表达谱的芯片。美国Stanford大学的David Botstein利用cDNA微阵列芯片,对乳腺癌细胞的基因表达进行了分析,发现其基因表达水平明显低于正常细胞。利用基因芯片对表达进行分析,在一次试验中可以获取相当于在60余万次传统的Northern杂交中所获得的关于基因表达的信息。通过这种实验方法,可以建立一种全新的肿瘤分类学方法,即依据每个肿瘤细胞中的基因表达情况对肿瘤细胞进行分类。基因芯片技术在分析基因的表达中具有不可比拟的优势。3.发现新基因 Moch等利用肿瘤微阵列芯片(5184个cDNA片段)发现了肾细胞癌的肿瘤标志物基因,并于正常细胞进行比较。在532份标本中检测到胞浆纤维Vimentin的表达基因,阳性率为51%~61%。追踪观察,有Vimentin表达的患者,预后极差。人类大量ESTs给cDNA微阵列提供了丰富的资源,数据库中400000个ESTs代表了所有人类基因,成千上万的ESTs微阵列将为人类基因表达研究提供强有力的分析工具。这将大大地加速人类基因组的功能分析。定量检测大量基因表达水平在阐述基因功能、探索疾病原因及机理、发现可能的诊断及治疗靶等方面是很有价值的。如该技术在炎症性疾病类风湿性关节炎(RA)和炎症性肠病(IBD)的基因表达研究中,由RA或IBD组织制备探针,用Cy3和Cy5荧光素标记,然后与靶cDNA微阵列杂交,可检测出炎症疾病诱导的基因如TNF-α、IL或粒细胞集落刺激因子,同时发现一些以前未发现的基因如HME基因和黑色素瘤生长刺激因子。Schena等人报道了cDNA的微阵列在人类基因表达监测、生物学功能研究和基因发现方面的应用。采用含1,046个已知序列的cDNA微阵列,用高速机器人喷印在玻片上,用双色杂交法定量监测不同基因表达,在一定的实验条件下,不同表达模式的阵列成分通过序列分析鉴定其特征。该方法较以往常用的方法敏感10倍以上,检测限度为1:500,000(wt/wt)总人体mRNA。在培养T细胞热休克反应的测定中,发现17个阵列成分的荧光比较明显改变,其中11个受热休克处理的诱导,6个呈现中度抑制,对相应于17个阵列成分的cDNA测序发现5个表达最高的成分是5种热休克蛋白,17个克隆中发现3个新序列。另外,在佛波酯诱导检测中,发现有6个阵列成分信号增强超过2倍,测序及数据库比较揭示有5个已知的,诱导表达最高的两个是PCA-1酪氨酸磷酸酶和核因子-κB1,有一个是未知的。这4个新基因的表达水平均相对较低,仅呈现2倍的诱导。Northern杂交结果证实了微阵列的结果。进一步检测了人的骨髓、脑、前列腺及心脏组织中热休克和佛波酯调节基因的表达,4种组织中检测出15种热休克和佛波酯调节基因的表达,其表达水平与Jurkat细胞中相应成分的表达水平密切相关如在四种组织中表达水平最高的两个基因β-actin和细胞色素C氧化酶在Jurkat细胞中的表达水平也很高。上述实验提示在缺乏任何序列信息的条件下,微阵列可用于基因发现和基因表达检测。目前,大量人类ESTs给cDNA微阵列提供了丰富的资源,数据库中400,000个ESTs代表了所有人类基因,成千上万的ESTs微阵列将为人类基因表达研究提供强有力的分析工具。这将大大地加速人类基因组的功能分析。4.大规模DNA测序 人类基因组计划的实施促进了高效的、自动化操作的测序方法的发展。芯片技术中杂交测序(sequencing by hybridization,SBH)技术及邻堆杂交(contiguous stacking hybridization,CSH)技术即是一种新的高效快速测序方法。用含65536个8聚寡核苷酸的微阵列,采用SBH技术,可测定200bp长DNA序列,采用67108864个13聚寡核苷酸的微阵列,可对数千个碱基长的DNA测序。SBH技术的效率随着微阵列中寡核苷酸数量与长度的增加而提高,但微阵列中寡核苷酸数量与长度的增加则提高了微阵列的复杂性,降低了杂交准确性。CSH技术弥补了SBH技术存在的弊端,CSH技术的应用增加了微阵列中寡核苷酸的有效长度,加强了序列准确性,可进行较长的DNA测序。计算机模拟论证了8聚寡核苷酸微阵列与5聚寡核苷酸邻堆杂交,相当于13聚寡核苷酸微阵列的作用,可测定数千个核苷酸长的DNA序列。Dubiley等人将合成的10聚寡核苷酸固定于排列在载片表面的0.1×0.1×0.02mm或1×1×0.02mm聚丙酰胺凝胶垫上制备聚寡核苷酸微阵列,先用分离微阵列(fractionation chips)进行单链DNA分离,再用测序微阵列(sequencing chips)分析序列,后者联合采用了10聚寡核苷酸微阵列的酶促磷酸化、DNA杂交及与邻堆的5聚寡核苷酸连接等技术。该方法可用于含重复序列及较长序列的DNA序列测定及不同基因组







 我要推广仪器
我要推广仪器
 下载APP
下载APP