由于单位一台分析仪需要将钢瓶纯氧气中烃类脱除然后进分析仪,查资料大多数都采用活性炭脱除,但是总感觉活性炭容易和氧气容易反应,不知道各位有没有这方面的经验,或者别的吸附剂?
序号】: 1【作者】:【期刊】:中国皮革【题名】:铬鞣剂中铁的脱除方法研究【年、卷、期、起止页码】:2007年9月【全文链接】:http://202.119.208.220:8002/kns50/detail.aspx?dbname=CJFD2010&filename=DWYX200903037
各位专家,大家好,目前我们遇到一个问题,在分析工艺回收氯苯时,其中含有微量的光气和氯化氢气体,ppm级,在进行色谱分析时,进样针不久就会被腐蚀,如针芯生锈,另外,检测器的收集极也会出现发黑的情况。不知道是否有遇到这种情况的,各位是如何解决这个问题的呢?是否有好的办法先脱除酸性物质而不影响待测组分的测定(待测组分怕水,怕胺、怕醇),希望高手指教,谢谢!
氨含量检测方法我公司对原料气中氨进行水洗脱除,现使用国产氨检测管检测,但对低浓度检测结果不满意。检测指标要求小于2.5ppm,现求检测气体中氨含量检测方法,请大家不吝赐教,谢谢。
请问那位朋友用于万通的[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]?是否配置了CO2脱除系统这个附件?对水质的分析CO2脱除系统是否有效提高灵敏度?
各位朋友,大家好!我在做农药残留分析时遇到一些问题,请大家帮帮忙! 我在净化的时候用的是自制的 5gFlorisil+2g氧化铝 柱,跟药典上的差不多,用的是 丙酮-正己烷(1:4) 洗脱液,可是洗下来的溶液还有颜色,柱子上吸附的不是很多, 请问这样净化后可以直接浓缩进样吗?会不会对 ECD检测器 有污染呢? 怎样才能有效脱除色素呢? 用活性炭时洗脱很慢,这是怎么回事呢?
各位大侠,求助啊!我做的是在惰性气氛下的实验,但是没有配套真空泵置换气体。实验过程中,通氩气十几二十分钟,发现仍有氧化的峰,说明氧没有置换干净。因为氧对实验的干扰很大,大家有没有好办法把仪器中的氧给脱出掉?氧的来源可能有二:一,由于没有真空置换仪器中的气体,通氩气不能把氧气置换干净; 二,氩气中含有微量的氧。
[color=#444444]最近要测有机染料(亚甲基蓝)里面的阴离子类型和大体含量,需要用到[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url],分析仪器的老师说预处理要去除液相中有机质,不然回去色谱柱有伤害,对实验有影响。求方法, 是要用萃取的方法么,用什么有机相来萃取能完全脱除?[/color]
空气中的硫化氢,大概30ppm,要求脱除硫化氢,排放空气达到国标要求,请问有什么好办法。空气流量为2500立方米/小时,装置要设计成多大?
测定水中阴离子浓度大于0.1ppm时,是否需配CO2脱除器?如用KOH体系不需CO2脱除器?
[color=#444444]在利用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]做催化剂活性测试时,由于气体组分中含有硫化氢,会对色谱有腐蚀,因此在进入色谱前需进行硫化氢脱除,该采取何种方法脱硫?查找的一些固体脱硫剂如氧化铁脱硫剂、氧化锌脱硫剂、活性炭脱硫剂等都是在大塔型设备中使用[/color][color=#444444],在净化反应管中无法完成脱硫任务,想请教各位大侠可知道适用于净化反应管中脱硫的脱硫剂?组分中含有二氧化碳,不能对其含量造成影响。谢谢啦![/color]
请问,分子筛可以用来脱除有机溶剂中的微量水分吗?无水氯化钙呢?
新买一根 SinoChrom ODS-BP 250*4.6 5um的柱子,装上之后用 乙腈(0.1%三氟乙酸):水=10%~100%的梯度洗脱 220nm和254nm下 出现鬼峰 1. 用90%乙腈 等度洗脱无鬼峰2 用50%乙腈等度洗脱也无鬼峰3用50%~90%的梯度洗脱出现鬼峰
虽说脱氧管可以有效地脱除氧气,但是受限于脱氧管再填充的困难性及其昂贵的价格,气体脱氧处理还是比较费力。极性柱子怕氧气,但是没有明确的数据说明氧含量低于多少的时候柱子是相对安全的,各位高手们是否有经验值呢?Agilent的脱氧管一般可以将载气中的氧气浓度降低至1ppb。另外如果用氢气作为载气的话,柱子对氧气的耐受程度是否会相对好一些呢?
我们这个项目是想利用脱气膜 的脱气功能脱出地下水中的O2、CO2、H2、He、CH4、H2S,脱气膜的填充材料是聚丙烯,纤维孔状,不知道会不会对H2S等气体产生吸附?请高人指点一二
本实验过程是想将自来水中的各种气体脱出,送入[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]进行检测,我们使用了两种脱气方法,一种是脱气膜装置,另外一种则是喷淋脱气装置,但两种装置实验过程中,在[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]上均未检测到氢气峰(偶尔有峰出现,但没有重现性),而其它自来水中的气体O2、CO2、N2、Ar2均能检测到,理论上说氢气应该是最容易脱出来的气体,不知道哪位大侠做过类似的实验,有高见能否分享?不胜感激!PS:用热真空脱气机脱气能检测到氢气,但我们以后想做在线脱气装置,所以考虑了喷淋与脱气膜装置,不知道还有没有更有效的在线脱气装置。
抑制器的in管不知道为什么脱出了,请问有什么方法可以重新接回去吗?[img]https://ng1.17img.cn/bbsfiles/images/2022/11/202211141044448678_3330_5382887_3.png[/img]
样品中有强保留的物质(高K’值)以馒头峰样被洗脱出,从而表现出一个逐步升高的基线,怎么解决?
样品中有强保留的物质以馒头峰样被洗脱出,从而表现出一个逐步身高的基线,该如何解决??
乙醇脱水产物有:乙烯,乙醚,乙醇,水。用哪种柱子能在[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]上测出这几种物质?
设置200结果脱出来了,所以流了很多液体出来,是不是压力太大,还是会不会哪里堵塞了呢,求高手帮忙
请问脱氧柱里面使用的可再生脱氧剂一般是什么材料呢,脱氧深度能达到多少呢?1ppm?
请问各位大侠,有谁做过无水乙醇脱水制乙烯在线分析,为什么我们的在线分析极不稳定,无法重复性,[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]需要注意哪些条件控制?反应系统压力与分析系统压力如何进行匹配?
各位行家们:请教个问题,我今天做乙烯利,质谱方法都调谐出来了,开始用UPLC分离的时候就老是拖尾,我用Atlantis T3 2.1*150mm柱子做分离,因为乙烯利是酸性物质,我就用甲醇:0.1%甲酸=10:90做,一直有拖尾,改用梯度也不行,后来用乙腈和甲酸做还是不行,各位大侠们,有没有好点的建议,急需!!!
老师们好,又有新问题请教。我用液相做梯度洗脱,流动相70%水:30%乙腈保持5分钟,10分钟内将乙腈升至100,再保持10分钟,结束。运行乙腈空白时,谱图上1.684min出峰,应该是溶剂峰。后面,14min--18min出现七、八个峰,有两、三个峰信号强度比溶剂峰大很多,差不多有十几倍。我不知道这是什么原因造成的,是我用的进口色谱纯试剂有问题,还是梯度设置的有问题?请教老师们了,帮我分析下出现这种情况还有什么原因。谢谢!
毕业临近,需要做一下聚氯乙烯/蒙脱土纳米复合材料的TEM样品制备,我对这方面不是很了解,请教其他人也没有知道的,在此请教高手给予指导。很着急,谢谢大家!
[size=5]色谱条件来自英国药典,梯度洗脱,平衡柱子后连续进两针水,分别如图:都有尖峰,并且保留时间,峰面积不同,重复性不好,这是为什么?两泵的流动相为:甲醇:水:乙腈:三乙胺,除三乙胺都是色谱纯,并且三乙胺1000ml中只加0.3ml,系统也没有气泡,平衡了一天,第二天还是这样。[/size]
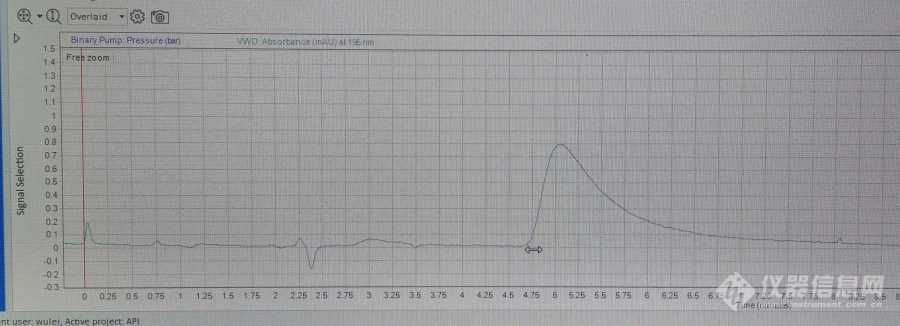
新手求助。欧洲药典关于环磷酰胺的有关物质方法变更[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相[/color][/url]方法,用了23%乙腈水,1.0流速等度洗脱,195nm。试着跑了空白溶液(同流动相)但是总是在5min出现大的鼓包。这是什么原因导致的呢?色谱柱用了不久,进了纯水也会出现这个包。[img]https://ng1.17img.cn/bbsfiles/images/2023/12/202312272138590270_7809_6347593_3.jpg[/img]
最近扩项,按照HJ/T 39-1999《 固定污染源排气中氯苯类的测定方法》——无水乙醇洗脱—气相色谱法,对于氯苯类的采样,需要一种很奇特的富集剂。标准原文如下:5.13 富集剂:二乙烯苯与乙基苯乙烯共聚物类多孔高分子小球型载体,比表面积约400㎡/g,颗粒度0.45-0.9mm。事先在脂肪提取器中用无水乙醇(5.1)处理8个小时。晾干后于80℃烘8h,备用采样用的富集柱:于40mm×5mm(内径)的硬质玻璃柱中,填装0.5g富集剂(5.13)并于两段塞少量玻璃棉,或视样品浓度,适当增加柱长度。
请问聚乙烯微珠如何通过热脱附进入GC-MS进行检测?







 我要推广仪器
我要推广仪器
 下载APP
下载APP