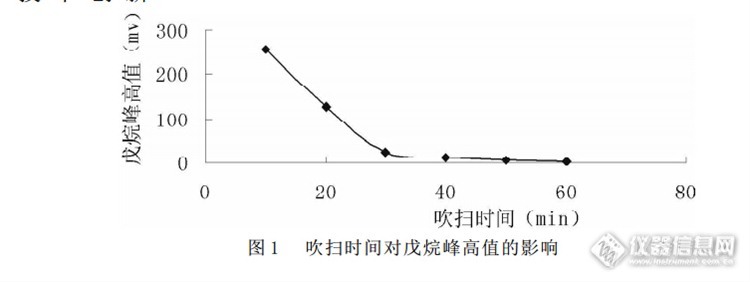
150℃)。戊烷作为非极性有机物,沸点仅 36 ℃, 可见在室温下, 戊烷是不易被Tenax-TA 吸附剂保留的物质在室温下, 分次注入戊烷 1.5 μ L, 洗脱气按不同时间段吹扫, 然后进行热解吸。从图 1 中看出: 室温下, 随着吹扫时间增长, 戊烷的峰高值逐渐降低。说明戊烷在活化过程中不被 Tenax-TA 吸附剂保留,而是通过缓慢释放而起作用的。戊烷分子在高温汽化后, 分子的热运动可能使高温下释放出的气体在戊烷分子和吸附剂之间建立了瞬间的气固相平衡。由于戊烷分子的不断流出, 平衡不断被打破, 而将其它杂质气体快速带出。戊烷分子由于自身的非极性和不被保留的性质, 而又逐渐被释放出来。3 结语利用气相色谱 - 热解吸仪器的吹扫功能, 选择戊烷作为 Tenax-TA 吸附管的洗脱剂, 在戊烷加入量为 1.5 μ L, 氮气流量为 80ml/min 时, 对吸附管活化 1h, 完全能够满足生产的要求, 本方法已应用于生产,成倍的提高了工作效率. 活化成本降低 50%以上。参考文献 室内空气质量标准.实施指南.中国标准出版社,112-120. 民用建筑工程室内环境污染控制规范.辅导教材. 中国计划出版社,281. 刘虎威. 气相色谱方法及应用. 化学工业出版社,167-168.
重量法蒸汽吸附仪 产品简介重量法动态蒸汽吸附仪DVS系列在测量水和有机蒸汽在粉体表面吸附方面处于世界领先地位,它通过在一定相对湿度下气体通过样品后重量的变化来测定蒸汽吸附,比传统的干燥法测量更快,更节省时间。由于其独特的优势,DVS系列产品世界各地的实验室有广泛的应用,可用于研发部门以及质控部门确定产品结构、产品稳定性、吸湿性、包装和产品开发中固体材料存在的问题。结合了微天平、气体流动和蒸汽的测量技术的优势使用干燥的载气,通常为氮气,可以选择任何两个蒸汽源中的一个质量流量控制和独特的水和有机蒸汽浓度实时监控结合可以精确控制饱和干燥载气流量的比例整个体系的温度可以由选择,并且在闭合环条件下可以精确控制,以保证吸附质的蒸汽压恒定具有极其高的灵敏度和精确度,仅需少量的样品(通常1-30mg),因而可快速达到平衡全自动惰气吹扫装置和有机泄露检测器可在发生有机蒸气泄漏时关闭联锁装置,保证安全 DVS Advantage软件可程序控制仪器,用户界面友好,满足数据完整性和安全性的最高标准待测样品置于微量天平上,已知浓度的蒸汽通过样品,记录式微天平可以测量由蒸汽吸附或脱附引起的质量变化。这种动态流动环境易于快速研究吸附/脱附过程。如果进一步实验选择需要,样品可以首先预热,这样可以加速体相吸附或者无机氧化物干燥过程的分析循环时间。加热过程可独立进行或通过软件来控制升温速率。
请教一下各位大神,Tenax吸附管最大吸附量大概是多(800左右的管),比如说最大吸附量达到100ug就会穿透,还有采样流量对吸附的影响有多大0.1L/min,0.2L/min....0.5L/min等,有哪位大神做过相关的探讨?是不是流量越大,吸附效果就越差?特别是对便宜的管来说。
我在室温和800度下分别做了脉冲吸附,八百度下的吸附量比室温下多,M脉冲吸附后吹扫升温至800度我又做了TPD,脱附量比吸附量还多,请问是怎么回事?
请教各位:活性炭的吸附能力怎样测试。我主要是想测试两种活性炭的吸附性能哪一种更好。我以前的方法是将活性炭放到墨水里面,静放一段时间后观察墨水的颜色变化。但是,我感觉这种方法太麻烦了。想请教各位有没有什么更好的、快速的方法测试两种活性炭的吸附性能?
用原子荧光做砷的测定时,用枪移标品还有样品,会不会被吸附?
扩项做7702.7,标准要求是对样品估值后按照0.01mol/L,0.02mol/L,0.03mol/L的滤液浓度,取三份样进行实验。我做完后发现取样多的反而小,取样少的反而吸附碘量多,这正常吗?按理来说不应该取样多吸附碘量多,取样少吸附碘量少吗?
谢谢。还是检测三氟乙酸的问题,我是要检测药品中三氟乙酸的残留量。如果柱子会吸附的话,是否意味检测出药品中的微量三氟乙酸?
在接80%醇洗脱液浓缩干燥后,发现茶多酚含量连80%都没有的到,会是哪里出了问题?请教各位大侠:为什么80%醇洗脱液液颜色很深,而且浑浊,尤其是一开始,似乎,一换成乙醇水,柱子上的东西全部下来了。即使先用20%的醇先洗1个柱体积,再换成80%醇,刚才说到的现象仍然没有改变。不知道这个现象是否正常?天气冷,洗脱液中乙醇比例是不是需要下降?还是我选择的非极性D101树脂不合适?最近我在用大孔吸附树脂纯化茶多酚时候,用的方法是:1. D101型大孔吸附树脂湿法装柱,2. 茶多酚粗品水溶后以0.8BV/h速度上样,3. 上样结束后会停止半小时,让样品充分和树脂接触吸附,4. 接下来用纯水快速过柱子2个柱体积,流速是2BV/h,5. 用80%的乙醇水洗脱,一般洗2个柱体积,流速为1BV/h.有经验的同学请多多指教!
在接80%醇洗脱液浓缩干燥后,发现茶多酚含量连80%都没有的到,会是哪里出了问题?请教各位大侠:为什么80%醇洗脱液液颜色很深,而且浑浊,尤其是一开始,似乎,一换成乙醇水,柱子上的东西全部下来了。即使先用20%的醇先洗1个柱体积,再换成80%醇,刚才说到的现象仍然没有改变。不知道这个现象是否正常?天气冷,洗脱液中乙醇比例是不是需要下降?还是我选择的非极性D101树脂不合适?用大孔吸附树脂纯化茶多酚的方法是:1. D101型大孔吸附树脂湿法装柱,2. 茶多酚粗品水溶后以0.8BV/h速度上样,3. 上样结束后会停止半小时,让样品充分和树脂接触吸附,4. 接下来用纯水快速过柱子2个柱体积,流速是2BV/h,5. 用80%的乙醇水洗脱,一般洗2个柱体积,流速为1BV/h.有经验的同学请多多指教!
我在室温和800度下分别做了脉冲吸附,八百度下的吸附量比室温下多,M脉冲吸附后吹扫升温至800度我又做了TPD,脱附量比吸附量还多,请问是怎么回事?
本人对于气体吸附量的测定方法不是很清楚,而且也是刚开始接触,主要想做多孔固体对气体的吸附性能,但目前对于如何才能较科学地测定固体对气体的吸附量尚缺乏有关的技术、知识。特向有过这方面经验的老师、先辈们请教!谢谢!e-Mail:zhyg98@163.com
请教去除食品中的油选择什么吸附剂?一般的吸附剂氧化铝、活性炭使用前怎么预处理?请大家说说自己的经验
14%。吸附剂更换填充比较: 双塔本身的体积很大,重新填充吸附剂的难度大,成本也高,并且重新填充的吸附剂在使用不久后又会出现吸附剂失效的问题。 模芯干燥机吸附系统的集成化设计,可实现现场快速更换吸附剂等问题,有效解决了原有吸附式干燥机现场更换吸附剂工作难度大、时间长及填充不紧密等严重缺陷。性能稳定性比较: 双塔干燥机采用体积庞大的两个压力容器罐体作为吸附模组,罐体内空间较大,导致气流分布不均匀。在进气口与出气口中间部分压缩空气的流量集中, 使中间部分的吸附剂过快饱和。饱和后的吸附剂无法有效的对压缩空气中的水分进行吸附,压缩空气夹带大量水分从中央集中通过,这种现象称为“隧道效应”。“隧道效应”造成处理效果不稳定,导致用气端有大量液态水。 模芯干燥机的新型吸附模芯由吸附管、吸附剂、扩散网板、密封材料和连接构件组成,采用高效暴风雪式填充技术,并通过高频振动专用模芯填充机进行灌装,吸附剂填充更为紧密,效果更干燥,性能更稳定。
请教各位充分干燥样品使表面吸附水分,这个量如何检测?
对于99.995%的高纯吸附载气和吸附质气体,其中的主要杂质气体为水份。假设气源气体中水份的含量为0.004%,则样品处在-195.8℃、30ml/min的流速中120min内停留在粉末表面的水的量为 0.14ml(标况下的体积),而对于500mg比表面积为1m2/g的材料,在其表面形成水的单分子层吸附所需要的水蒸汽的量为:0.12 ml(标况),与实际停留在粉末表面的水量相当,材料表面已经被水分饱和;如果不吹扫处理继续测试,那测试结果将不可能正确。对于色谱法孔径测试需要测试三四十个分压点,影响更是显著,若分压点之间不做吹扫处理,最后得到的结果将不是固体材料本身对氮分子的吸附了,而是包覆了水分子的颗粒对氮分子的吸附了,孔隙也早已被高沸点易吸附气体杂质H2O、CO2饱和。 要消除吸附质气源中的气体杂质H2O、CO2等的影响, 可采用冷阱气体净化装置,冷阱是消除高沸点气体杂质的有效方式;比表面仪配备的冷阱,使本会被样 品吸附的水份等高沸点杂质提前被冷阱捕获,使得经过净化后的高纯氮和高纯氦气体中的水分含量低于10-17Pa,达到超高纯气体状态; 3H-2000系列比表面仪是国内唯一配备冷阱的比表面仪器,这也是该系列仪器能够取得高精度和高分辨率的因素之一。
大家好,请问吸附-脱附曲线应该怎么分析?从图中可以分析出什么东西呢?在下新手,能否请各位老师专家尽量讲的详细点?这两个样品都是用酸处理方法制备的多孔玻璃,只是工艺参数不同。测试仪器是ASAP2020。谢谢大家。
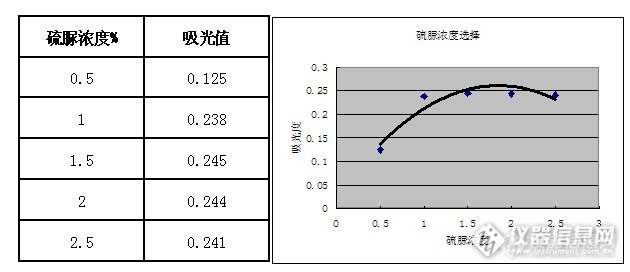
泡塑吸附—火焰原子吸收光谱法测定矿石中金含量【摘 要】本文主要采用泡塑吸附-火焰原子吸收光谱法测定矿石中金含量,对溶液酸 度、吸附时间、解析时间、硫脲浓度等条件进行探索,得出优越的实验条件。【关键词】泡塑吸附 火焰原子吸收光谱法 金 条件选择 金在自然界含量极低,根据最新研究成果,金的地壳丰度值仅为1.1×10-3g /t。随着金矿的普查勘探,对金的分析提出了更高的要求。目前,金的富集方法很多,主要有铅试金法、活性炭吸附法、溶剂萃取法、泡沫塑料吸附法等; 金的测定方法也很多,主要有重量法、氢醌容量法、碘量法、硫代米蚩酮光度法、原子吸收法、等离子体质谱法等。自20 世纪70 年代发现泡沫塑料对金属元素有吸附性以来,由于这一新的富集技术简便易行、成本低廉,因而得到快速发展,并广泛应用于金的富集分离。本文采用王水溶矿-泡沫塑料富集-硫脲解脱-火焰原子吸收法测定金,并在实验的基础上优化了测定条件。采用硫脲解脱,避免了因灼烧引起的空气污染,方法操作简便,干扰离子少,精密度高,测定结果稳定,重现性好。该方法应用于大量地质样品中金的测定,结果令人满意。1、实验部分1.1 仪器与试剂仪器:原子吸收分光光度计(普析,TAS990)、马弗炉、锥形瓶(250ml)、比色管等试剂:盐酸、硝酸、1.0mg/ml金标准储备液1.2 仪器工作条件http://ng1.17img.cn/bbsfiles/images/2013/09/201309051621_462593_2352694_3.jpg1.3 实验方法1.3.1样品制备(详见帖子http://bbs.instrument.com.cn/shtml/20130706/4835569/)1.3.2称取25.0g金矿样于蒸发皿中,放入马弗炉中630度恒温1h(升温到630度需1h,共两小时);(详见帖子http://bbs.instrument.com.cn/shtml/20130711/4845138/)http://ng1.17img.cn/bbsfiles/images/2013/09/201309051627_462596_2352694_3.jpg1.3.3放冷后转入250ml锥形瓶中,用少量水润湿,加入50ml王水(1:1),在电热板上加热使样品溶解,溶液剩余10ml左右取下,冷却,用水冲洗瓶壁,并加水至120ml左右。1.3.4每个锥形瓶中加入两块准备好的海绵,震荡40min,取下。1.3.5每块海绵用水冲洗30次,放入加有25.00ml的1%硫脲的比色管中,水浴20min。http://ng1.17img.cn/bbsfiles/images/2013/09/201309051628_462597_2352694_3.jpg1.3.6尽快取出海绵,放冷后待测。1.4 标准曲线绘制用1.0mg/ml金标准贮备液逐级稀释至10.0ug/ml金标准工作液。移取一定量的金标准工作液,配置一系列金标准工作系列溶液(浓度及移取体积见下表)按照1.3.4-1.3.6步骤处理标准溶液,并绘制标准曲线。http://ng1.17img.cn/bbsfiles/images/2013/09/201309051623_462594_2352694_3.jpghttp://ng1.17img.cn/bbsfiles/images/2013/09/201309051624_462595_2352694_3.jpg2、结果讨论2.1加入王水酸度的选择移取金标准工作溶液10.0ml5份于250ml锥形瓶中,溶液酸度分别控制在10%,15%,25%,30%,35%,分别加入两块泡塑,于震荡机上震荡40min,取出泡沫塑料,用自来水冲洗干净; 将泡沫塑料放入25 mL 比色管中,加入1.0 %硫脲的溶液25 mL,置于水浴锅中解脱20 min; 取出比色管,捞出泡沫塑料,挤干。待溶液冷却后,以原子吸收光谱法测定。[/size
动态水分吸附法是一种非常适合分析材料水分吸附性能和记录水分吸附等温线的检测方法,适用于粉末,颗粒,碎片、片剂或块状固体。吸附仪常用来进行新材料的稳定性测试,这种长时间的测试可能需要几天、几周甚至是几个月,能够为评估环境温湿度对产品保质期产生的影响提供非常有价值的数据。 更进一步来说,分析研究在某一温湿度条件下有多少水分能够透过包装渗透到内部被材料本身吸附非常重要,被吸附的水分从外界环境中迁移到包装内部是影响带包装物体保质期的主要原因。 采用动态水分吸附仪来检测带包装药品或食品的水蒸气吸附性能,对于产品防潮性的检测和保质期的预测有着重要的指导意义。
介绍 环境、生物药物、食品和香料中的有机物的分析通常需要将待测物从基质(饮用水、废水、体液、饮料等)中提取和富集。目前大多数样品前处理方法包括液-气萃取或平衡法(冲洗和收集,顶空分析),液-液萃取或固相萃取。 过去几年里,微型化已成为分析化学的一个主要趋势。样品前处理方法微型化的典型事例包括微量液-液萃取(瓶内萃取),室温静态顶空和盘式固相萃取。通过与先进的分析仪器联用,在保证或提高检测灵敏度的前提下,这项技术实现了更快的分析速度,更高的样品通量、较低的溶剂消耗、较低的劳动力花费。几十年前,Arthur和Pawliszyn 提出了一种新的微萃取的方法,即固相微萃取(SPME)。80年代中期,不同研究小组分别报道了采用涂有PDMS薄膜的开管柱收集阱,以聚二甲基硅氧烷作为萃取介质对含水样品或气相中有机物进行萃取的实例。以PDMS作为介质的萃取是基于物理吸着而不是化学吸附作用。如Baltussen等所述,吸着性浓集与吸附过程相比具有多种优点。这些优点包括浓集效果可以预测,不存在转移效应,吸附材料化学性质稳定,可在较温和条件下快速解吸。但是,实际应用中的局限(低载样量,低上样体积……)限制了PDMS涂层开管柱收集阱的应用。另一方面,SPME是一种使用十分简便、快捷的技术。在针的外层涂有一薄层PDMS膜(7-100μm)作为萃取介质。吸附完成后,化合物在GC进样器中热解吸或在LC进样器中进行液体解吸。与PDMS涂层的开管柱收集阱不同的是,SPME本质上是一种相平衡技术,该技术基于溶质在硅氧烷相及水相分配行为的差异进行提取。近来研究发现,这一平衡与溶质在辛醇/水中的分配系数(KO/W)有关。这些研究表明,当溶质的KO/W较低(KO/W10000)时,其回收率也较低,这主要是由于水相和PDMS相两相间的相比较大产生的。SPME中PDMS的用量常常只有0.5μl或更少,因此限制了样品在PDMS纤维上的富集量。基于上述研究,近年来开发出了一种新的使用PDMS涂层搅拌棒进行萃取的方法。在这种方法中,PDMS的用量为50-300μl,因此,检测灵敏度增加了100到1000倍。当溶质的KO/W大于500时,可获得100%的回收率。当溶质的KO/W在10到500之间时,可采用SPME相同的方法对分析结果进行校正。这项技术称为搅拌棒吸附萃取(SBSE)。
请教诸位大侠,吸附氨的灰样需先经过滤,请问用什么过滤方法好呢?只担心,吸附的氨不会完全溶解出,过滤不干净。谢谢大侠们关注!煤灰在高温时(大概800-1000摄氏度)吸附了氨,现在想检测其吸附的氨含量是多少?谢谢!
据我所知可吸附卤素含量是检测废水中的指标,但是我的客户却要求在有机产品中检测可吸附卤素的含量,需要特定的仪器,帮个忙,介绍一下哪里能测?
活性炭碘吸附值达到多少为一级品合质金的分析方法都有那些?用湿法,[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]测定可以么?干法有成熟的方法么?
[align=center][font=DengXian]搅拌棒吸附萃取[/font]SBSE[font=DengXian]特点[/font][/align][font=DengXian]对于样品基质复杂,其香气风味成分测定需要一种简单快速,无溶剂或少许溶剂的提取富集技术。搅拌棒吸附萃取[/font]SBSE[font=DengXian]具有灵敏度,操作简便特点。和一般[/font]LLC[font=DengXian],[/font]SDE[font=DengXian],[/font]SPE[font=DengXian],[/font]SAFE[font=DengXian]等样品提取制备方法相比,搅拌棒吸附萃取([/font]SBSE[font=DengXian])是一种无溶剂的用于萃取和浓缩痕量有机物的技术。不需要大量溶剂,样品量少,无需浓缩等步骤。是一种绿色无溶剂化学分析方法。[/font]SBSE[font=DengXian]具有比[/font]SPME[font=DengXian],以及[/font]SPME Arrow[font=DengXian]更大的吸附层体积([/font]24-126 [font=DengXian]μ[/font]L vs. 0.5 [font=DengXian]μ[/font]L vs. 10.2 [font=DengXian]μ[/font]L[font=DengXian]),故其灵敏度比[/font]SPPME[font=DengXian]要高出[/font]50-250[font=DengXian]倍,同理也比[/font]SPME Arrow[font=DengXian]要高几倍。图[/font]1[font=DengXian]为[/font]SBSE[font=DengXian]、[/font]SPME Arrow[font=DengXian]、[/font]SPME[font=DengXian]的理论回收率的示意图,当样品体积为[/font]10mL[font=DengXian],[/font]SBSE[font=DengXian]的萃取层体积为[/font]24 [font=DengXian]μ[/font]L[font=DengXian],[/font]SPME Arrow[font=DengXian]的萃取层为[/font]10.2[font=DengXian]μ[/font]L, SPME[font=DengXian]的萃取层为[/font]0.5[font=DengXian]μ[/font]L[font=DengXian]时,对不同极性(以[/font]Log Ko/w[font=DengXian]指数来表现)的化合物的理论回收率。[/font]
我们最近采用气质检测1,2-二氯乙烷含量,采用的顶空法。做曲线还可以,可是检测样品的时候发现未检出。加标回收回收率很低。样品前处理很简单,就是1g样品加10ml水,放在20ml顶空瓶中,80度水浴加热,然后顶空进样,进样量100ul.后来查了一些资料,发现我们的样品,是一种吸附性材料,可以吸附二氯乙烷,我想问一下大家有什么好的办法来检测样品中本身的二氯乙烷呢?
[em06] 用的康塔的Autosorb-1做Pd/HZSM5的化学吸附 方法是500度下H2还原1小时,40度下H2吸附问题:测得的结果表明随着Pd担载量的增加,Pd的颗粒越来越小,分散度(有人说用表面暴露度更好)越来越大,这个结果和一般的结果是矛盾的。用同一个方法做了一系列的8个样品,结果都是这个规律很奇怪,不知道哪里出错了,因为H2吸附时候的溢流造成的吗?虽然说用CO吸附更好一点,但是也有人用H2做Pd的吸附啊会是哪里出问题了呢?求救求救
误将吸附作用当做的仪器故障 有这样一个真实的经历,或许是因为没有经验而引起的,现在拿出来跟大家分享一下。那是我刚学会气相色谱法没多久,想着独立检测一批比较复杂的试验,但是在做的时候突然发现不管是对照品还是样品,只有溶剂峰,我们所用的溶剂是二甲亚砜。不出峰是挺伤心的,也挺紧张的,害怕会耽误生产。然后抓住一个同门师兄帮忙解决问题,结果还是一样的,都没有出峰。后来,我们从头到尾逐一排查,先是从进样口,密封垫换新的,衬管拆下来看,感觉没什么问题,石英棉看起来比较干净,而且上次用了还是没问题的(后来查找原因时发现可能是因为石英棉上有吸附剂而导致不出峰),就认为衬管也没有问题。色谱柱也是跟衬管一样的想法,上次使用时没有问题,所以也是相当然的觉得没有问题,只是为了排除各种可能,重新拆卸后再重新安装,结果还是一样。最后就是检测器,将喷嘴拿去用甲醇超声都没有效果。也认为是仪器收到污染了,但是不管进的纯的溶剂还是纯的标准品,都没有太好的效果。因为是新手,所以还不是很清楚应该如何处理,最后暂且将问题总结为检测器的问题,就想这试试ECD,正准备去查找资料的时候,进了一针异丙醇(这个是用于清洗检测器用的),等了大概20分钟左右,结果奇迹居然发生了,之前怎么都不会出峰的居然突然出峰了。好吧,真不知道什么原因,就抱着试试的态度,进了一次混标,好吧,我承认这次真的是瞎蒙,乱搞,居然搞对了,好吧,我只能说我是瞎猫撞到死老鼠,死马当活马医,居然收到奇效!总结一下,导致不出峰的原因可能是吸附作用,吸附的位置应该在石英棉上。查找了一下仪器使用台账,果然是因为吸附而导致的(出于保密原则,就不说吸附物质的名称了),但不会吸附在色谱柱上。其实真正吸附于何处应该无从验证了,因为当时的经验上的缺乏,对仪器不熟悉,所以不会去仔细分析,不会想到更换衬管,还是一味的寻找检测器的问题。最后想说一下,经过后来更多的试验后积累了一些经验,试验失败有各种可能,只针对一个怀疑对象,很难找出其中真正的原因所在。后来还有一次,试验失败了,以为是一起的问题的,通过查找资料,最后得出结论,是前处理的问题,所用的试剂达不到相应的要求(氢碘酸浓度低于57%),浓度达不到要求,就导致反应不充分,对于含量的检测,当然就不准确了。当然,实际情况实际分析,不能一出问题就盲目的从源头查找,例如有一次,进样后突然有一针不出峰,当时排查了很久,就是找不到原因,结果经过工程师的提醒,确认是自动进样针堵了,更换进样针之后问题解决。在做实验遇到问题时,应多向有经验的人请教,或许他也不懂你的问题到底出在哪,但是经过分析,结合你做实验的过程,自己都可以发现问题所在。例如有一次,升级了系统之后,按照原来的程序设置,一切都没有问题,只是色谱柱的信息需要重新输入,当我将正确的色谱柱信息输入后,按照以前的方法进行操作,结果发现出峰时间延后了(因为是程序升温,所以运行时间是固定的),在这做必要的说明,因为我们是做质检的,需要严格的按照程序设置程序,所以不会增加运行的时间。整个程序运行完毕之后,还有两个峰没有分离完成,降温过程中出峰了,属于异常情况,根据平时的经验,每个环节都检查一遍,发现没什么问题,结果请教工程师,其实工程师也不知道是什么原因,将怀疑的地方都检查了一遍,没发现问题。最后通过一次很随意的讨论发现,原来是因为我将流量设置错误而导致的。用一句话概括就是,用原来本应该是错误的方法(也不能说是错误的,只是相对于而言)来应用于一个正确的色谱柱信息(以前的色谱柱信息就没对上号)上,从而导致一个失败的试验发生。以上是我的一些经验总结,或许很简单,很多大师都觉得不应该出现这样的低级错误,但是偏偏我遇到了,作为一个新手,我只能说我太笨了,好吧,就这样,谢谢大家。
有做吸附方面的吗?比如说吸附水中重金属,等有害物质?吸附方面不大了解,想请教一下通常的吸附机理有freundlich和langmuir,大多数吸附时符合其中一种机理,要么是freundlich,要么是langmuir,但是我在测活性炭吸附的时候,遇到同时符合这两种机理,是不是有这种情况啊,还是我哪里出问题了,有相关的资料吗?
SN/T 1958-2007 进出口食品中伏马毒素B[sub]1[/sub]残留量检测方法 酶联免疫吸附法2007-08-06发布,2008-03-01实施,现行有效。[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=137208]SN/T 1958-2007 进出口食品中伏马毒素B1残留量检测方法 酶联免疫吸附法[/url]
文献上用亚甲基蓝脱色力来衡量活性炭的能力,想问一下甲基蓝脱色力是不是就是标准上说的甲基蓝吸附量?如果不是,那它是什么?具体怎么算一直弄不明白谢谢了







 我要推广仪器
我要推广仪器
 下载APP
下载APP