推荐厂家
暂无
暂无
 留言咨询
留言咨询
留言咨询
留言咨询
 银牌2年
银牌2年
 400-860-5168转6028
留言咨询
400-860-5168转6028
留言咨询
 400-886-5615
留言咨询
400-886-5615
留言咨询
 400-838-7877
留言咨询
400-838-7877
留言咨询
 400-860-2797
留言咨询
400-860-2797
留言咨询




[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url]的几种定量分析方法[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=44491][url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url]的几种定量分析方法[/url]
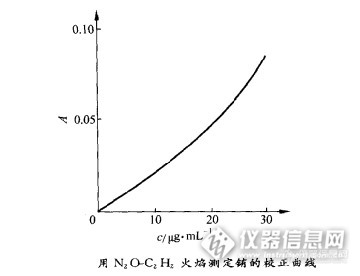
影响原子吸收光谱定量分析的因素原子吸收光谱定量分析涉及两个基本过程:①试样中被测元素转化为自由原子的化学过程;②蒸气相中自由原子对辐射吸收的物理过程。化学过程比物理过程更复杂,影响化学过程的因素比影响物理过程的因素更多。1 原子化过程的影响在推到原子吸收光谱定量分析的关系式A=Kc时,假定了一个基本条件:在确定的实验条件下,蒸气相中的原子数N与试样中被测元素的含量c成正比,N=βc,为此要求被测元素的原子化效率在确定的实验条件下是一定的。准所周知,在实际分析工作中所遇到的试样类型千变万化,即使是同一元素,在不同的试样内,由于基体特性各异和其他共存元素的相互影响,其原子化效率各有不同,有时甚至差别很大。原子化效率对实验条件非常敏感,在原子吸收这类高温动态测量中,实验条件的变动性导致原子化效率的改变,几乎是不可避免的。这是影响原子吸收光谱分析的准确度和精密度的主要因素。由此可以得出这样的结论,测定一种试样中某一元素的最佳条件,未必适用于另一种试样中同一元素的测定,必须针对具体分析对象,寻求某一元素测定的最佳条件。现在商品原子吸收光谱仪器中,厂家为用户所提供的预先储存在数据库内各元素的分析条件,多半都是用纯溶液样品得到的,只能作为选择实际分析样品分析条件的参考。计算机的广泛使用、原子吸收仪器自动控制系统的日益完善以及横向加热石墨炉和STPF技术的应用等,为获得稳定的原子化条件提供了可能性。化学过程是一个复杂的过程,有关影响化学过程的因素。2 辐射吸收过程的影响从光源的发射线考虑,在原子发射线中心频率V0的很窄的△V频率范围内,kv随频率的变化很小,可以近似地认为kv→k0,。当空心阴极灯光源的发射线远小于原子吸收线的宽度时,如下图所示,测得的吸光度可以近似地认为是峰值吸光度。http://ng1.17img.cn/bbsfiles/images/2015/11/201511161143_573671_2352694_3.png随着空心阴极灯的灯电流增大,由于自吸和多普勒变宽效应增强,使光源发射变宽,对于低熔点金属Cd,Zn和Pb等元素空心阴极灯,光源发射线和原子吸收线宽度几乎达到同一数量级,使测得的峰值吸光度明显地降低,导致校正曲线严重弯曲。下图使用不同灯电流时所得到的镉校正曲线。http://ng1.17img.cn/bbsfiles/images/2015/11/201511161144_573672_2352694_3.png在入射辐射中,若含有非吸收辐射,如连续背景辐射、空心阴极灯内稀有填充气体与支持材料以及其他杂质发射的辐射等,它们都可能出现在光谱通带内。当不存在非吸收辐射时,吸光度A=lgI0/I,当存在非吸收辐射i0时,吸光度A’=lg(I0+i0)/(I+i0),A’小于A0。i0在整个入射辐射中所占比例越大,A’比A小的越多。i0和I0比例一定时,I值越小,即吸收介质内分析原子浓度越高,i0的影响越大。非吸收辐射i0的存在,使测得的吸光度减小,校正曲线弯曲。从吸收谱线轮廓考虑,在通常的原子吸收光谱分析条件下,分析原子浓度都很低,共振变宽效应可以忽略不计。但是,当吸收介质的分析原子浓度高时,同种分析原子相互碰撞引起谱线共振变宽,使峰值吸光度减小。随着分析原子浓度增大,对峰值吸光度的影响增大,因此,造成校正曲线在高浓度区弯向浓度轴。这是导致校正曲线非线性化的重要因素。在建立峰值吸收的定量关系式http://ng1.17img.cn/bbsfiles/images/2015/11/201511161141_573669_2352694_3.png时,假定吸收谱线轮廓主要由多普勒变宽效应决定。事实上,吸收谱线轮廓不仅受多普勒变宽效应的影响,还与碰撞变宽,特别是洛伦茨变宽有关。在有些情况下,多普勒变宽与洛伦茨变宽是同一数量级,不能忽略其影响。洛伦茨变宽还引起吸收谱线轮廓的频移与非对称化,使得测定的吸光度不能代表峰值吸收,而是中心波长两侧的吸光度,其值低于峰值吸光度,导致校正曲线的非线性化。谱线的精细结构是影响吸光度测量的又一可能的因素。这些相差很小的谱线精细结构常常是简并的。对于很重和很轻的元素,其波长差超过了线宽,在这种情况下,测定的吸光度是精细结构内各组分的混合吸光度,而非单一纯组分的吸光度,故导致校正曲线的弯曲。当用锐线光源进行峰值吸收测量时,谱线的精细结构对吸光度测定的影响可以忽略不计。下表列出了某些元素共振线的同位素移值。http://ng1.17img.cn/bbsfiles/images/2015/11/201511161145_573673_2352694_3.png从吸收介质内原子浓度考虑,在推到吸收关系式http://ng1.17img.cn/bbsfiles/images/2015/11/201511161142_573670_2352694_3.png时,认为入射辐射密度pv是不变的。很显然,只有在吸收层很薄或分析原子浓度很低时才是这样,这说明原子吸收光谱法主要用于痕量和超痕量元素分析。当被测元素的浓度高时,引起吸光度下降,校正曲线弯向浓度轴。由此可知,原子吸收光谱分析的校正曲线线性范围不会很宽,一般是1-2个数量级。在通常的原子吸收条件下,可以忽略激发态原子和元素电离的影响,但对于低电离电位元素,特别是在高温下,不能忽略电离对基态原子的影响。电离度随温度升高而增大,在一定温度下,随元素浓度增加而减小。元素电离的影响如下图所示,电离效应导致校正曲线弯向纵轴。http://ng1.17img.cn/bbsfiles/images/2015/11/201511161145_573674_2352694_3.png
[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]理论知识--定量方法[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url]法是一种元素定量分析方法,它可以用于测定60多种金属元素和一些非金属元素的含量。定量分析方法:一、标准曲线法:配制一系列不同浓度的待测元素标准溶液,在选定的条件下分别测定其吸光度,以测得的吸光度A为纵坐标,浓度为横坐标作图,得到标准曲线。再在相同条件下测定试液的吸光度,由标准曲线上就可求得待测元素的浓度或含量。注意事项:1.配制标准溶液时,应尽量选用与试样组成接近的标准样品,并用相同的方法处理。如用纯待测元素溶液作标准溶液时,为提高测定的准确度。可放入定量的基体元素。2.应尽量使得测定范围在T=30~90%之间(即A=0.05~0.5),此时的测量误差较小。3.每次测定前必须用标准溶液检查,并保持测定条件的稳定。4.应扣除空白值,为此可选用空白溶液调零。二、 标准加入法: 取两份体积相同的试样溶液,设为A和B,在B中加入一定量的待测元素,然后分别将A和B稀释到相同体积,再分别测定其吸光度。设A中待测元素的浓度为Cx吸光度为Ax,B中的待测元素浓度为Cx+Co(Co为加入的标准样品的浓度),吸光度为A,则:Ax=KCx A=K(Cx+Co)两式相比得:Cx=Co×Ax/(A-Ax)由此式就可得到待测元素的含量。作图的方法 :取四份以上的体积相同的试液,从第二份开始,分别按比例加入不同量的待测元素,将这些溶液全部稀释到相同体积,此时,各溶液中待测元素的浓度分为:Cx,Cx+Co,Cx+2Co,Cx+3Co等测定各溶液的吸光度,并以吸光度对加入的待测元素的浓度(增量)作图,得如下曲线:? 将直线延长至与横坐标相交,交点与原点之间的距离所代表的浓度值就是试液中待测元素的浓度。 注意事项:须线性良好;至少四个点;只消除基体效应,不消除分子和背景吸收;斜率小时误差大。










 我要推广仪器
我要推广仪器
 下载APP
下载APP