药物稳定性实验箱和药物光照实验仪的区别 ,如果药物稳定性实验箱提供0-8000的光照 是否还需要光照实验箱
抗菌药沙星类抗菌药是光毒反应发生率较高的一组,很多名称中都带有“沙星”两个字,如环丙沙星、氧氟沙星、依诺沙星等等,属于抗菌药的喹诺酮类别。据统计,沙星类抗菌药导致光毒反应发生率为0.1~3%,服用该类药物皮肤经光照以后,常见的症状是皮肤红肿、发热、瘙痒、疱疹等。 除杀星类抗菌药物外,还有四环素类、磺胺类、美满霉素、多西环素、地美环素等抗菌药,也有光毒反应的报道。 抗精神病药如氯丙嗪,在临床上,它是用来治疗精神病、镇吐、低温麻醉及人工冬眠等情况的药物。氯丙嗪为处方药,因此在服药前可找专科医生或药师咨询相关情况,服药后也要做好防护,如戴墨镜及避免皮肤直接暴露于阳光下等。 利尿药如速尿、双氢克尿噻(氢氯噻嗪)等,也就是能增加排尿的药物,也是高血压疾病常用的治疗药物,在水肿、心衰、胸水、腹水的也多用到该类药物,临床上也有其发生光毒反应的报道。 抗组胺药如扑尔敏、苯海拉明等均为第一代康组胺药,常用于抗过敏治疗医|学教育网搜集整理。 解热镇痛药如布洛芬、阿司匹林等,很多人对该类药物都不陌生,在感冒、疼痛、消炎时,都可能会用到,在用药日晒后,若出现皮肤反应时,要加以警惕。 降糖药如格列苯脲、格列吡嗪等,该类药物也是用量较大的药物,它们均为处方药,若在服药后出现光毒反应时,应及时找专科医生咨询或换药,切不可擅自更改或停药。 抗肿瘤药:如长春新碱等,长春新碱常用于治疗急性及慢性白血病、恶性淋巴瘤、小细胞肺癌及乳腺癌等,为静脉注射药物,并不是非处方药。
大家好,我现在想用生化培养箱代替药物稳定性试验箱,做长期稳定性试验,不知道可不可行,据我了解,生化培养箱都有很好的控温控湿功能,并且有光照,而且价格比药物稳定性试验箱便宜,请问大家,生化培养箱长期运转会不会有问题,哪位有这方面的使用经验,分享一下。谢谢啦。
大家好。我是一名新手,近日要采购一台光照试验箱,要求符合ICH Q1B要求和中国GMP的强光试验要求。但是在选择设备的时候,不知如何选择。同时也包括采购设备后的使用过程,如何操作能够满足以上光照试验要求?比如:UV的波长怎样确认?光照试验时,使用ID65与UV同时照射?还是分开照射?光的照度多少LUX更合适?要求不低于1200000LUX.HR。在采购光照箱时,如何确认与验收与检测ID65与UV 的光强度?对于ID65与UV二种光源是必须要做二种试验吗?我计划做试验的产品是:醋酸甲地孕酮,醋酸甲羟孕酮,屈螺酮等产品。请各位老师指导谢谢。
[font=-apple-system, BlinkMacSystemFont, &][size=15px][color=#05073b] 植物光合作用测定仪反应灵敏度高吗,植物光合作用测定仪的反应灵敏度通常是非常高的,这主要得益于其先进的传感器技术和设计。以下是一些关于植物光合作用测定仪反应灵敏度的详细信息和特点: 传感器技术: 植物光合作用测定仪配备了高精度的传感器,用于测量与光合作用相关的关键参数,如二氧化碳浓度、空气温湿度、叶片温度、光照强度等。 这些传感器通常具有快速响应能力,能够迅速捕捉到微小的环境变化,并准确地转化为数据输出。 测量精度: 由于采用了高精度的传感器和先进的测量技术,植物光合作用测定仪能够提供非常准确的测量数据。 例如,一些光合作用测定仪的二氧化碳测量精度不会受到温度变化的影响,并且具备稳定、高精度、反应灵敏等特性,可以在一秒钟以内完成二氧化碳差值收集。 智能化系统: 许多植物光合作用测定仪配备了智能化系统,能够实时显示、储存和传输测量数据。 这种智能化系统可以大大提高测量的便捷性和效率,同时也能够确保数据的准确性和可靠性。 稳定性: 光合作用测定仪通常具有良好的稳定性,能够在长时间连续测量中保持高灵敏度。 这对于需要进行长时间监测或连续监测的研究项目来说尤为重要。 多功能性: 植物光合作用测定仪可以同时测量多个参数,如光合速率、蒸腾速率、细胞间二氧化碳浓度、气孔导度等。 这种多功能性使得它能够满足不同研究项目的需求,并提供全面的数据支持。 综上所述,植物光合作用测定仪的反应灵敏度通常是非常高的。它采用了高精度的传感器技术、先进的测量技术、智能化系统和稳定的设计,能够迅速、准确地捕捉到与光合作用相关的微小环境变化,并提供准确的测量数据。这些特点使得植物光合作用测定仪在植物生理学、生态学、农业科学等领域的研究中具有重要的应用价值。[img=,690,690]https://ng1.17img.cn/bbsfiles/images/2024/06/202406131144468576_457_6098850_3.jpg!w690x690.jpg[/img][/color][/size][/font]

[b]药品稳定性试验箱[/b]采用了温度、湿度、光照等综合性环境模拟方式,来对药品进行温度、湿度、光照等条件下的稳定性。对于制药行业来说,药品稳定性试验箱是必不可少的药品检验试验设备,根据其检测药品保存的温度、湿度、光照强度等情况来了解药品的很佳保存方式,以及药品的稳定性检测。[align=center][img=,348,348]https://ng1.17img.cn/bbsfiles/images/2021/07/202107291711294462_7879_1037_3.jpg!w348x348.jpg[/img][/align] 药品稳定性试验箱利用温度、湿度、光照环境来对药品的失效条件和时间进行检测,尤其是对一些新药进行加速试验、高温试验和强光照试验,这种方式是目前制药企业对药品的稳定性进行检测的很佳方式。 综合药品稳定性的作用有两种,一是采用高温、低温、高低温交变、空气湿润、强光照等多环境因素来对药品的稳定性进行测试,根据试验测试情况来对药品进行改善,以得到稳定性很强的药物 二是对于一些特殊药物进行检测,找到其保存的很佳环境因素。目前药品稳定性试验箱主要是用来检验药品在环境因素下的稳定性。 通过药品稳定性试验箱的测试,可以获得关于药品对温度、湿度、光照条件下的测试数据,可以了解该药品在环境因素下稳定性变化情况,然后根据这些数据制药企业可以确定药物生产的包装、储存条件和安全的有效使用期限。
紫外-可见分光光度法在药物分析中的应用郑 健 陈焕文 刘宏伟 李宝华 张寒琦 于爱民 金钦汉(长春分析仪器研究和技术开发中心,吉林大学化学系分析教研室,长春,130023)摘 要:本文着重评述了1998年以来国内紫外-可见分光光度法在药物分析中的应用,展望了其在该领域的发展趋势。关键词:紫外-可见分光光度分析法 药物分析 综述中图分类号:O657.32 R917 文献标识码:A 以分子吸收光谱为基础的紫外-可见区分光光度分析法具有设备简单、适用性广、准确度和精密度较好等特点,已在地质[1]、环境[2]、能源[3]、材料[4]、食品[5]等科学中发挥着重要作用,尤其是随着多元络合物、胶束增敏光度法、有机试剂等的发展,它已经成为应用最广泛的分析手段之一[6]。分光光度法的早期应用集中在无机分析化学领域,即对为数众多的无机离子和化合物进行定性分析或定量测定[7]。 有机物的光度分析较无机物的开展晚,但发展十分迅速,已经从定性、半定量分析发展到定量分析,方法的灵敏度和选择性也有了很大的提高,能够用光度法进行分析测定的物质种类也在不断增多[8-10]。近几年来,随着生命科学的发展,有机物光度分析的研究和应用热点主要集中在生物[11]、临床、药物[12]等方面。光度法在药物分析中的应用历来受到大家的广泛关注和重视,世界各国都进行这一领域的相关研究[13,14]。据统计,在药物分析中,分光光度法占29.1%,色谱法占25.5%,荧光、化学发光法占2.4%,与光度法有关的方法共计占31.5%,由此可见,光度法是药物分析最常用的方法之一[12]。研究结果表明,光度法在药物分析中的可靠性可以和色谱法相蓖美[15],但其设备简单、操作方便、价格低廉、易于普及等特点是色谱法难于做到的。因此,可以相信,光度法在药物分析中将大有作为。 我国学者在药物光度分析领域开展了大量工作,取得了喜人的成果。建立了紫外-可见分光光度法[16]、发光光度法[17]、荧光光度法[18]等许多新的测量方法和体系[12,19],测定的药物涉及生物碱、苯磺酰胺、芳胺、芳烃、巴比妥、甾体激素、咪唑、噻吩、吡啶、季胺盐、磺酰胺、氯胺酮类、醇、醛、醚、某些杂环化合物、某些糖及苷、抗生素和维生素等几大类。本文只评述1998~2000年间紫外-可见分光光度法在药物分析中的应用和进展,在药物分析中的其它光度分析方法,如荧光光度法[20-22]、发光分析[23],可以参阅相应的近期综述。
想找一台有三种波长的光照试验箱,其中一种是360nm左右的,箱子里面有十几根灯管,各自分三种波长,可控制用不同波长灯管做光照实验,不知谁家做
如题,请问有用过的版友赐教,顺便问下重庆永生的药品强光照射试验箱SHH-100GD的价格是多少!谢谢了!
大家好,请问能否给我一些关于药物稳定试验的有关英文的资料?谢谢。我现在急需要。

紫外老化试验箱是一款光老化测试机器设备,实验关键对于于建筑涂料、面料、塑胶等商品。给大家做一个紫外光老化试验箱的光照冷疑系统软件的分析。让顾客更掌握这款实验设备。 首先是它的光照系统软件,[url=http://www.linpin.com/][b]紫外老化试验箱[/b][/url]标准配置8支led灯管(led灯管选用Q-LAB,使用寿命1600h上下),设备启动led灯管默认设置打开情况,即做光照实验(仿真模拟大白天自然光中的紫外光对商品的脆化全过程),它能够和自动喷淋系统相互配合应用(光照时另外自喷),加重老化测试标准。光照时实验室溫度一般设置在65℃之内,光照時间、自喷時间、实验总時间都可以依据产品执行标准设置必须時间。[align=center][img=,690,690]https://ng1.17img.cn/bbsfiles/images/2022/07/202207191700383276_1915_1037_3.jpg!w690x690.jpg[/img][/align] 必须留意的是,国内紫外线灯管使用寿命一般为600h上下,当led灯管使用期来到总使用寿命三分之二时,就必须拆换led灯管了,由于此刻led灯管光波长早已衰减系数,达不上标准的实验光波长标准。 也有一点,光照温度仪表和冷疑温度仪表2个仪表盘的溫度是不可以另外显示信息的,有顾客就担心过这个问题。为什么不另外显示信息呢?由于一个表明的是大白天做实验的溫度,一个表明的是夜里做实验的溫度,大白天和夜里是不可以另外存有的,因此这两个溫度是依据设置实验時间更替显示信息的。 紫外老化试验箱冷疑系统软件是仿真模拟夜里露珠,粘附在商品表层对商品的浸蚀老化状况,这时候光照不是工作中的。它还可以和自动喷淋系统另外应用,便是仿真模拟夜里下雨天的实验标准。冷疑和自动喷淋系统由箱身体的水泵压力开关和储水箱内的2个离心水泵操纵其渗水。保证设备在做实验的情况下有充足供电。冷疑溫度一般设置在50℃~55℃以内,自然顾客能够依据自身商品的规范去设置溫度。
大家好,请问能否给我一些关于药物稳定试验的有关英文的资料?谢谢。我现在急需要。
维权声明:本文为012304原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现的,均属侵权违法行为,我们将追究法律责任。本实验主要考察实验过程中时间、光照、以及温度对不稳定药物在样本基质中的变化;从而使检测得到的数据结果更加的可靠;本实验以己烯雌酚为目标药物,鸡肉、鸡肝为样本基质;进行验证;实验基础样本种类:鸡肉、鸡肝药物类型:己烯雌酚检测手段:酶联免疫试剂盒检测方法检测限:0.5ppb药物浓度:空白添加3ppb 药物环境条件实验室温度:15-20度 湿度:20-30%样本添加方式:将一大份样本分为两份,将药物溶解后以喷雾的行洒在已经匀浆的样本中;进行多次混匀,并检测该样本和等条件下没有添加的另外半份样本(除了没有添加药物外其他条件完全相同);实验设置条件设置如下:避光 太阳光直射 37度温箱 正常条件下2小时 1小时 1小时 30min6小时 2小时 2小时 30min1天 3小时 3小时 30min2天 4小时 4小时 30min试剂/药品(以下药品均为分析纯级别)乙腈、丙酮、三氯甲烷、磷酸、氢氧化钠(分析纯),去离子水前处理方法鸡肉样本的处理过程称取2+-0.05g样本到40ml塑料离心管中,加入6ml乙腈-丙酮(80:20),振摇10min,离心后取3ml吹干,取3ml到玻璃试管中,于40度水浴氮气吹干,加入0.5ml三氯甲烷涡动溶解后,再加入2ml M氢氧化钠溶液,涡动30s,离心后去上层1ml,加入200ul 6M磷酸,混合后加入6ml乙腈,振荡5min,离心后取全部上层吹干,并加入1ml溶解进行溶解;鸡肝的前处理处理过程称取2+-0.05g样本到40ml塑料离心管中,加入6ml乙腈-丙酮(80:20),振摇10min,离心后取3ml吹干,取3ml到玻璃试管中,于40度水浴氮气吹干,加入0.5ml三氯甲烷涡动溶解后,再加入2ml M氢氧化钠溶液,涡动30s,离心后去上层1ml,加入200ul 6M磷酸,混合后加入6ml乙腈,振荡5min,离心后取全部上层吹干,并加入2ml溶解进行溶解;以下是鸡肉样本的数据闭光放置时间 2小时 6小时 1天 2天空白值 0.21 0.16 0.27 0.17检测值 2.93 2.80 2.71 2.45回收率 98% 93% 90% 82%37度时间 1小时 2小时 3小时 4小时空白值 0.20 0.15 0.24 0.21检测值 2.96 2.46 2.21 1.81回收率 99% 82% 74% 64%阳光照射时间 1小时 2小时 3小时 4小时空白值 0.14 0.19 0.17 0.16检测值 2.81 2.31 1.92 1.74回收率 94% 77% 64% 58%正常条件下 30min空白值 0.13检测值 2.89回收率 96%鸡肝的数据如下闭光放置时间 2小时 6小时 1天 2
1.前言 致癌试验的目的是考察药物在动物体内的潜在致癌作用,从而评价和预测其可能对人体造成的危害。任何体外实验、动物毒性试验和人体应用中出现的潜在致癌性因素均可提示是否需要进行致癌试验。国际上,对于预期长期使用的药物已经要求进行啮齿类动物致癌试验。在研究药物的潜在致癌作用中,致癌试验比现有遗传毒性试验和系统暴露评价技术更有意义。这些试验也可帮助理解无遗传毒性药物的潜在致癌作用。目前常规用于临床前安全性评价的遗传毒性试验、毒代动力学试验和毒性机理研究的数据,不仅有助于判断是否需要进行致癌试验,而且对于解释研究结果与人体安全性的相关性也是十分重要的。由于致癌试验耗费大量时间和动物资源,只有当确实需要通过动物长期给药研究评价人体中药物暴露所致的潜在致癌性时,才应进行致癌试验。 2.历史背景 在日本,根据1990年“药物毒性研究指导原则手册”,如果临床预期连续用药6个月或更长时间,则需要进行致癌试验。尽管连续用药少于6个月,如果存在潜在致癌性因素,也可能需要进行致癌试验。在美国,大多数药物在广泛应用于人体之前,已进行了动物致癌试验。根据美国药品食品监督管理局(FDA)要求,一般药物使用3个月或更长时间,需要进行致癌试验。在欧洲,“欧共体药品管理条例”规定了需要进行致癌试验的情况,包括长期应用的药物,即至少6个月的连续用药,或频繁的间歇性用药以致总的暴露量与前者相似的药物。 自2005以来,我国《药品注册管理办法》附件中规定预期临床连续用药6个月以上或需经常间歇使用的药物应进行致癌试验,并指出了进行致癌试验的多个考虑因素。2007年1月国家食品药品监督管理局药品审评中心发布的《治疗用生物制品非临床安全性技术审评一般原则》中阐述了相关产品致癌试验的要求。 2009年10月药品审评中心组织毒理专家、企业和研究单位代表召开了制定“药物致癌试验必要性技术指导原则”专题讨论会,会上基本认同了ICH S1A中内容的适用性,并结合国内情况进行了一些调整。
[b][url=http://www.f-lab.cn/vivo-imaging/msot.html]小动物光声成像系统[/url][/b]MSOT是全球唯一能够提供[b][url=http://www.f-lab.cn/vivo-imaging/msot.html]小动物全身光声成像[/url][/b]能力的小动物实时光声成像系统,用于临床前小动物成像和临床前研究。小动物光声成像系统能够可帮助生物过程和药理物质作用在体内,在深部组织中高分辨率下实时观察。小动物光声成像系统是全球唯一混合光声超声成像技术,OPUS成像技术的同类仪器,也是世界上第一个交叉断层成像系统,提供非平行的用户独立的图像质量,并且具有实时性,可以获得整个动物的横截面影像。这套小动物光声成像系统包含组织形态基于血红蛋白信息产生的光声层析成像,反射式超声成像的集成(r-uct)能力添加互补的解剖信息,特别是低灌注结构。小动物光声成像系统可以调谐激发激光波长,采集光声信号,执行多个波长的光谱分解,这样内源性色基团以及外在探针可有效被区分。小动物光声成像系统工作MSOT探测器小动物置台可以利用各种手持探测器实现小动物的二维和三维自动成像。动物置台可作为内部图像和EIP MSOT成像系统的附件。主要特点包括:自动数据采集三维阶段控制加热的动物垫激光安全联锁装置动物监控摄像机接入导管或生命体征监测[img=小动物光声成像系统]http://www.f-lab.cn/Upload/MOST-invision-imaging.JPG[/img]小动物光声成像系统混合光声超声成像技术(OPUS成像)小动物光声成像系统是全球唯一混合光声超声成像技术,OPUS成像技术的同类仪器,也是世界上第一个交叉操作断层成像系统,提供非平行的用户独立的图像质量,并且具有实时性,可以获得整个动物的横截面影像。这套小动物光声成像系统包含组织形态基于血红蛋白信息产生的光声层析成像,反射式超声成像的集成(r-uct)能力添加互补的解剖信息,特别是低灌注结构。[img=小动物光声成像系统]http://www.f-lab.cn/Upload/Hybrid-OPUS-IMAGING.jpg[/img]初步实验表明,小动物光声成像系统t的升级版将应用在以下需要可视化的任何结构:肿瘤边缘转移胰腺膀胱小动物光声成像系统技术信息单波长的光声成像在10 Hz帧频高达5赫兹帧频的实时频谱分量可视化公司注册的反射式超声计算机断层扫描(r-uct)MSOT IN VISION 512-ECHO成像穿透深度2-4厘米,适合全身小动物成像。横截面的空间平面分辨率:150μM高功率/快速可调谐激光系统(100兆焦耳/ 10毫秒)具有64/128/256/512元件的断层超声探测器阵列全自动图像采集用于光谱和时间分析的数据后处理套件[b][/b]
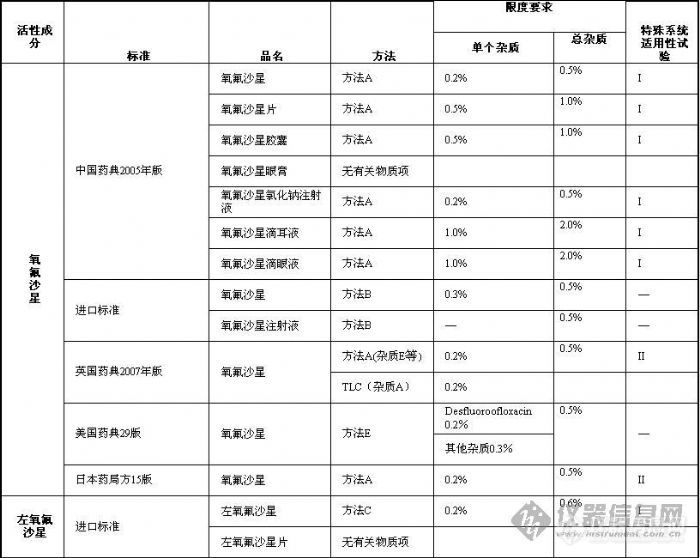
作者 张立雯 成海平 正文内容 【摘要】本文总结了国家标准中氧氟沙星、左氧氟沙星、盐酸左氧氟沙星、乳酸左氧氟沙星、甲磺酸左氧氟沙星系列药物的有关物质控制方法,分析了该类药物注册申报中有关物质控制存在的问题,希望能为研发者提供帮助。 【关键词】氧氟沙星、左氧氟沙星、有关物质一、概况 氧氟沙星(Ofloxacin)为合成的第三代广谱氟喹诺酮类抗菌药,对大多数革兰氏阳性菌和革兰氏阴性菌均有明显的抑制作用。临床上主要用于敏感菌所致的呼吸系统感染、泌尿生殖系统感染。氧氟沙星由日本第一制药株式会社研发,于1985年在日本、德国上市,制剂为口服片剂、注射剂等。目前国内已上市的氧氟沙星制剂有片剂、胶囊剂、颗粒剂、缓释制剂、小针、葡萄糖注射液和氯化钠注射液等。 左氧氟沙星(Levofloxacin)为氧氟沙星的左旋体,具有抗菌谱广、抗菌作用强的特点。日本第一制药株式会社于1993年在日本上市销售左氧氟沙星原料及片剂,并现已在英国、美国等多国上市。目前国内上市的左氧氟沙星制剂主要有片剂、小针、葡萄糖注射液和滴眼剂等。另外,国内已批准上市的左氧氟沙星还有其盐酸盐、乳酸盐和甲磺酸盐,三种加酸根的左氧氟沙星均有片剂、胶囊剂、注射制剂等多种剂型上市。二、国家标准中有关物质控制方法比较 氧氟沙星系列药物的有关物质测定国家标准大多采用HPLC法,列表比较见表1。 表1 氧氟沙星系列药物的有关物质测定方法与限度的比较 这些方法有很多相似的地方,如均采用ODS柱,色谱条件与含量测定色谱条件相同,按照主成分自身稀释对照法定量等。但也有一些不同的地方值得关注,作者从以下三个方面来对这些国家标准方法的不同之处进行比较。1、流动相 按照流动相的不同,作者将有关物质测定方法分为六种,具体如下: 方法A:醋酸铵高氯酸钠溶液(取醋酸铵4.0g和高氯酸钠7.0g,加水1300ml使溶解,用磷酸调节pH至 2.2)-乙腈(85∶15)为流动相,在294nm下检测; 方法B:略; 方法C:略; 方法D:己烷磺酸钠[取己烷磺酸钠0.98g,加磷酸盐缓冲溶液(取磷酸二氢钾6.8g,加水溶解并稀释至 1000ml,加0.05mol/L磷酸约500ml,使pH为2.4)]-甲醇(3∶1)为流动相,在293nm下检测; 方法E:磷酸缓冲溶液(溶解27.2磷酸二氢钾在1000ml水中,用磷酸调节pH至2.4)-乙腈(90∶10)为流动相,在294nm下检测; 方法F:己烷磺酸钠[取己烷磺酸钠0.98g,加磷酸盐缓冲溶液(取磷酸二氢钾6.8g,加水溶解并稀释至 1000ml,加0.05mol/L磷酸约500ml,使pH为2.4)]-甲醇(65∶35)为流动相,在230nm下检测。 六种流动相的共同特点是:组成均是酸性缓冲溶液加有机溶剂(甲醇或乙腈)。方法A、B、E未加表面活性剂,方法C、D加有表面活性剂己烷磺酸钠。除方法F在230nm下检测外,其他方法均在294或293nm下检测。2、主要杂质 英国药典收载了氧氟沙星杂质A、B、C、D、E、F共6个已知杂质,依次分别为去哌嗪环、去羧基、去氟、氟取代位置不同、去甲基以及氮氧化的化合物物。英国药典氧氟沙星原料药采用TLC法控制杂质A,采用HPLC法控制其他已知和未知杂质。美国药典重点关注了杂质desfluoroofloxacin,日本药局方重点关注了ofloxacin demethyl substance,均与英国药典的杂质E相同,是氧氟沙星的去甲基化合物,该化合物为氧氟沙星的主要降解产物,光照下极易产生。美国药典给出了desfluoroofloxacin的相应因子为1.13。中国药典没有明确已知杂质,但有关物质检查时采用以下光照降解法进行特殊系统适用性试验,光照试验中产生的杂质即为氧氟沙星、左氧氟沙星的主要杂质。 美国药典中desfluoroofloxacin按加校正因子的主成分自身稀释对照法定量,美国药典氧氟沙星其他杂质和其他国家标准中氧氟沙星或左氧氟沙星所有杂质按不加校正因子的主成分自身稀释对照法定量。有关物质限度的要求详见表1。3、特殊系统适用性试验 氧氟沙星、左氧氟沙星及其盐的含量测定和有关物质检查方法的系统适用性试验除通常的进样精密度、记录时间、理论塔板数等的要求外尚有一项较为特殊的系统适用性试验,其他标准方法采用I法,日本药局方采用II法,详述如下: 特殊系统适用性试验I(光照降解法):取供试品溶液于无色试管中,用日光灯(2500lux或3500lux)或紫外灯(254nm)照射1小时或3小时或4小时,取此液注入液相色谱仪,记录色谱图,相对保留时间约为主峰1.2处应能检测出色谱峰。 特殊系统适用性试验II(杂质对照品法):氧氟沙星和杂质E(ofloxacin impurity E CRS,英国药典)或氧氟沙星的去甲基物(ofloxacin demethyl substance)分离度不得低于2.0或2.5。 由于缺少杂质对照品,国内氧氟沙星系列药物的有关物质测定系统试验常常是采用光照降解法。也正是因为缺少杂质对照品,系统适用性试验才显得尤为重要,是考察系统分离能力的重要指标。 另外,左氧氟沙星及其盐的原料和制剂均需检查右旋异构体,方法基本相同,均是采用硫酸铜-L异亮氨酸溶液-甲醇或硫酸铜-D苯丙氨酸溶液-甲醇为手性流动相检测,限度要求不得过0.8%或1.0%。在此就不详加讨论。三、注册申报中存在问题与探讨1、不重视系统适用性试验 氧氟沙星系列药物的特殊系统适用性试验常常被忽视,其实却非常重要。若不做该项试验,就不能保证所采用的系统能将最难分离的相对保留时间1.2倍的色谱峰分离出来,就有可能得到错误的结果。 审评中曾发现申报盐酸左氧氟沙星注射液的某厂家自测盐酸左氧氟沙星含量较药检所检验结果高约5%(含量测定色谱条件与有关物质检查一致)。仔细审查其图谱,发现未按盐酸左氧氟沙星注射液的已有国家标准用光照降解法进行系统适用性试验,且色谱峰明显拖尾。其测定结果偏高很可能是紧随主峰之后的杂质峰包裹进了主峰。 申请人往往会留意进样精密度、理论塔板数这样的常规系统适用性试验,却常常忽略了光照降解系统适用性试验,此种现象在申报资料中占很大比例。究其原因,是试验人员没有理解到此项系统适用性试验的目的和重要性,希望提醒申请人提高对系统适用性试验的重视程度。2、没有杂质个数与含量的详细对比 申报资料中杂质对比研究通常的做法就是按照国家标准方法检验一下自制品和已上市对照药品,若都在标准规定范围内,就认为自制品与已上市药品质量相当。其实这样的做法是对杂质对比的目的和比什么不甚明了的表现。杂质对比一方面要了解自己的产品与已上市品杂质有哪些不同,另一方面要了解制剂过程中有没有新产生的杂质,若有新产生的杂质,应加以控制。所以比较就要落到列表对比杂质的个数与含量上,泛泛地比较杂质总量是不足以说明问题的。 例如,申报盐酸左氧氟沙星、乳酸左氧氟沙星的注射剂采用方法D测定的较为多见。在该色谱条件下,相对保留时间为0.23、0.43、1.2左右的杂质峰较常见,其中相对保留时间为1.2的色谱峰是稳定试验中含量有所增加的主要杂质。 另外,统计杂质的个数时要注意“忽略限度”。英国药典氧氟沙星有关物质项下明确规定:Disregard any peak with an area less than 0.1 times the area of the principal peak in the chromatogram obtained with reference solution (a),即忽略面积小于对照溶液主峰面积0.1倍的色谱峰(0.02%)。中国药典没有这么详细的规定,但从实际操作来看,为增强方法的严谨性和数据的可比性,建议申请人在统计杂质个数时应明确忽略限度。3、强力破坏试验降解程度不合适 有关物质检查方法学验证的重要项目就是通过强力破坏试验考察方法的专属性,但强力破坏试验破坏程度的掌握不尽合理。较容易出现的情况是破坏太轻微,酸、碱、氧化、光照破坏均几乎未产生可检测的杂质,这样就无法判断所采用的色谱条件分离能力是否符合要求,破坏试验失去意义。另一种极端是破坏过度,主峰降解了大半,产生大量重叠的杂质色谱峰,这样很难找到合适的色谱条件将所有的降解产物分离。个人认为,适度的破坏应是采用比贮藏中可能遇到的最强条件稍强烈的条件降解,产生比贮藏中可能产生的稍多杂质,若所选色谱条件能将这些杂质都分离,就是专属性符合要求的。 以上问题是氧氟沙星系列药物审评中经常遇到的问题,希望能为研发者提供帮助,共同努力提高我国仿制药的研发水平。 [img]http://ng1.17img.cn/bbsfiles/images/2009/07/200907202227_160694_1612824_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2009/07/200907202231_160696_1612824_3.jpg[/img]
注射剂如何进行药包材与药品相容性试验?最近在做注射剂更换药包材工作,要求做相容性试验。查看了一些相关资料,通常都是做影响因素与稳定性试验。但在进行咨询的时候,有人告知,现在只做这些应该是不行的了,要求做迁移、吸附以用添加剂对药物影响试验。因为完全没有经验,所以不知道以上实验应该怎么开展,希望有经验的朋友可以帮忙解答。谢谢!例如迁移试验,如何测定重金属的迁移呢?至于吸附试验,我个人理解,是不是测定一下药物含量,与最初测定含量相比较,无明显变化,是否就能说明药包材对药物无吸附呢?
一直以来,大型制药公司研发药物的临床试验数据都作为公司的高度机密,而保存在公司的数据库中。葛兰素史克(GSK)决定打破常规,公开公司的“高度机密”。讨论公布在研药物所有临床试验数据利弊.
第一章 总则第一条 为加强药物Ⅰ期临床试验的管理,有效地保障受试者的权益与安全,提高药物Ⅰ期临床试验的质量,根据《中华人民共和国药品管理法》、《药品注册管理办法》、《药物临床试验质量管理规范》等相关规定,参照国际通则,制定本指导原则。第二条 本指导原则适用于药物Ⅰ期临床试验,旨在为药物Ⅰ期临床试验的组织管理和实施提供原则性指导。人体生物利用度或生物等效性试验可参考本指导原则。第二章 职责要求第三条 申办者应建立筛选国家药物临床试验机构和研究者的程序和标准,选择、委托获得相关专业资格认定的药物临床试验机构进行药物Ⅰ期临床试验。 第四条 申办者应建立质量保证体系,对临床试验的全过程进行监查和稽查,确保临床试验的质量,保障受试者的权益与安全。申办者可以委托合同研究组织执行临床试验中的某些工作和任务。申办者对临床试验的真实性及质量负最终责任。第五条 国家药物临床试验机构的药物Ⅰ期临床试验研究室负责药物Ⅰ期临床试验的实施。研究者应遵循《药物临床试验质量管理规范》,并严格执行临床试验方案,对临床试验的真实性和质量负责,保护受试者的权益与安全。第六条 临床试验生物样本分析应在符合国家有关管理规定的实验室进行。第七条 伦理委员会在审查试验方案和知情同意书等资料的基础上,应针对药物Ⅰ期临床试验的特点,重点关注试验风险的管理及控制、试验方案和知情同意书的修改等,对临床试验进行必要的监督检查,加强对受试者的权益与安全的保护。
关于“天然药物及仿生药物”等国家重点实验室主任和学术委员会主任聘任的通知 教技司61号 有关高等学校: 根据科学技术部和财政部《国家重点实验室建设与运行管理办法》的有关规定,为保证实验室持续稳定发展,规范和加强对重点实验室领导班子聘任管理,经有关高校公开招聘和推荐,最终确定了“天然药物及仿生药物”等国家重点实验室主任的推荐人选和学术委员会主任建议人选。 经研究,我司同意聘任周德敏教授、陈凯先院士等分别担任天然药物及仿生药物等国家重点实验室主任和学术委员会主任(具体名单见附件),任期至下一轮评估终止。 附件:国家重点实验室主任和学术委员会主任聘任名单序号实验室名称依托单位实验室主任学术委员会主任1天然药物及仿生药物北京大学周德敏陈凯先2生物反应器工程华东理工大学许建和欧阳平凯3草地农业生态系统兰州大学南志标盖钧镒4环境模拟与污染控制清华大学黄霞曲久辉5病毒学武汉大学吴建国陈焕春6家蚕基因组生物学西南大学夏庆友向仲怀7机械结构强度与振动西安交通大学王铁军杜善义8农业生物技术中国农业大学李宁朱玉贤9植物生理学与生物化学中国农业大学武维华许智宏

[align=center][b][img=,600,336]https://ng1.17img.cn/bbsfiles/images/2019/09/201909121439522763_1873_932_3.jpg!w690x387.jpg[/img][/b][/align][b]质量研究与质量标准质量研究[/b]🔥 原料药的质量研究合成多肽原料药的质量研究除参考一般化学药物的研究思路进行常规项目的研究外,还应根据合成多肽的结构特征、制备工艺特点和生物学特点等进行针对性的研究,研究项目一般包括:外观性状、理化常数、鉴别、氨基酸组成分析、水分、反离子含量、纯度、有机溶剂和反应试剂残留量、生物学安全性检查、含量和/或活性效价测定等。检测方法研究和验证的基本思路和要求与已颁布的相关技术指导原则相一致。对于合成多肽药物,除常规项目外,理化常数一般需要关注其比旋度、等电点(pI)、溶解性(主要为水和缓冲液中)等。一般而言,多肽药物的常规检查项目与其它化学药物相同。此外,与多肽药物的结构及合成特点相关的一些检查项目,例如氨基酸组成分析、反离子(例如三氟醋酸或醋酸根)含量、反应试剂残留量(例如从树脂上裂解多肽使用了氢氟酸,需要检查氟化物残留量)等,则需要在原料药质量研究中予以重视。相关肽检查(或称有关物质检查)是反映多肽化学纯度的重要指标之一,根据多肽的理化性质、分子大小,可选择合适的色谱、电泳等方法进行。短肽可参考一般化学药品有关物质检查的研究思路选用适宜的方法;长肽的有关物质检查方法除常见的RP-HPLC外,还可考虑使用高效离子交换色谱(HPIEC)、毛细管电泳技术等,非解离条件下的高效分子排阻色谱(HPSEC)、聚丙烯酰胺凝胶电泳(PAGE)以及激光散射粒度测定等技术可用于聚合体/低聚体的检查。有关物质检查的方法学验证应能证明所采用的方法可以有效分离目标多肽与工艺杂质(例如缺失肽等)、降解产物(例如二硫键交换或氧化产物等)、聚合物等。一般应考察两种以上不同原理的方法,高效液相色谱法至少应包括一种梯度洗脱方法,并采用多肽粗品和强制降解试验等对方法的专属性等进行考察、对比,此外还应注意研究多波长检测的结果并选择合适的检测波长等。合成多肽因结构特征不同于通常的小分子化学药品,纯度检查有时难以从根本上有效控制产品安全性,需要进行必要的生物学安全性检查(如过敏试验、降压物质、升压物质、异常毒性等)以全面控制产品质量、保证安全性。此外,根据产品具体情况,对于长肽,有时尚需进行免疫原性或抗原活性等生物特性的研究。含量测定是评价多肽质量的重要指标之一,理化方法测定其含量时称为“含量测定”,生物学方法或酶化学方法测定其效价时称为“效价测定”。对于短肽,理化方法测得的含量可以反映其有效程度时,首选简单、通用的含量测定方法;对于具有一定空间结构才能发挥其活性的多肽,需进行生物学方法或酶化学方法测定药物活性(效价)的研究,包括含量与活性的关系、相应的方法学验证等。🔥 制剂的质量研究合成多肽制剂的质量研究基本思路和要求可参照《化学药物质量标准建立的规范化过程技术指导原则》、《化学药物制剂研究基本技术指导原则》等相关内容,根据合成多肽的具体特点,在原料药质量研究的基础上,结合剂型特点、处方工艺以及临床使用特点,重点研究所用辅料和制剂工艺对产品质量的影响、制剂辅料和制剂产生的降解产物对检测方法的影响以及与剂型相关的质量要素。研究项目一般亦应包括性状、鉴别、检查(安全性、均一性、纯度要求与有效性指标等)、含量或效价测定等几个方面。[b]质量标准[/b]合成多肽药物质量标准的制订原则、要求与《化学药物质量标准建立的规范化过程技术指导原则》是一致的。即,在系统的质量控制研究基础上,充分考虑药品安全、有效、质量可控的要求,以及生产、流通和使用等环节的影响,确定能够揭示、控制药物内在品质的检测项目、分析方法和限度要求,如原料药质量标准应包括氨基酸组成、等电点、中长肽的肽图等。合理可行的质量标准应能有效控制产品质量以保证临床用药的安全性和有效性,并有效地控制药品批间质量的一致性。相关质控项目的限度确定也应参考相关的指导原则,例如对于有关物质检查限度的确定可以参考《化学药物杂质研究的技术指导原则》、仿制品种同时还可参考《化学药品仿制研究技术指导原则》等的原则性要求,并结合产品本身的特性及临床使用情况,视具体情况而定。随着药物研发进程的深入,研究数据积累的不断丰富、方法学研究的完善和药物研究技术的不断发展,质量标准在不同研究阶段需要不断修订和完善。[b]稳定性研究[/b]合成多肽药物稳定性研究的基本原则应遵循《化学药物稳定性研究技术指导原则》的一般性要求。与一般化学药物相比,多肽药物的稳定性较差。引起多肽药物不稳定的原因主要有水解、氧化、外消旋化、二硫键的断裂及重排、β消除、凝聚、沉淀、吸附等。当多肽处于溶液中或高湿下保存时,其降解或聚合的速度会比干燥条件下大为增加。因此,稳定性研究应根据多肽药物稳定性的特点合理选择试验条件、考察项目。加速试验和长期留样试验的试验条件应依据药物对温度、湿度和光照等条件的敏感程度的考察(影响因素试验)基础上选择;考察项目除常规项目(例如原料药的比旋度、有关物质和含量等)外,根据具体情况,可能还需要考察其生物活性的变化。与其他化学药物不同,多肽药物可能具有一定程度的表面活性,有与直接接触药品的包装材料和容器发生吸附等相互作用的可能,从而引起制剂效价、生物活性下降。例如有些多肽分子能够与玻璃表面的硅醇基发生相互作用。因此,在包装材料的选择方面需注意其与多肽药物相互作用的研究,有些情况下可选择特殊处理后的包装容器,如表面经硅烷化处理的容器等。[b]名词解释非天然氨基酸:[/b][color=#717070]除自然界生物体中存在的氨基酸外,其它由人工合成制备的氨基酸。[/color][b]反离子:[/b][color=#717070]和多肽形成离子对的带有相反电荷的离子。[/color][b][b][/b][/b]参考文献1.Guidance for Industry for the Submission ofChemistry,Manufacturing,and Controls Information for Synthetic Peptide Substances,FDA,1994。2.合成多肽专题研讨会会议纪要,药品审评中心,2001。3.多肽药物分析方法研究进展,叶晓霞,俞雄,中国医药工业杂志,2003,34(7)。[b]著 者《合成多肽药物药学研究技术指导原则》课题研究组。[/b]
求助 :最新化学药物稳定性技术指导原则与药物质量试验控制分析验测实务全书,电子版,谢谢
推荐:实验室普通生物光学显微镜[img]https://ng1.17img.cn/bbsfiles/images/2023/06/202306302054141735_1341_5389809_3.jpeg[/img][img]https://ng1.17img.cn/bbsfiles/images/2023/06/202306302054144492_4607_5389809_3.jpeg[/img]
国食药监注483号各省、自治区、直辖市食品药品监督管理局(药品监督管理局),总后卫生部药品监督管理局: 为加强药物Ⅰ期临床试验的管理,有效的保障受试者的权益与安全,提高药物Ⅰ期临床试验的研究质量与管理水平,根据《中华人民共和国药品管理法》、《药品注册管理办法》和《药物临床试验质量管理规范》等有关规定,国家局组织制定了《药物Ⅰ期临床试验管理指导原则(试行)》,现予印发。请你局组织本行政区域内药物临床试验机构学习,参照执行。 附件:《药物Ⅰ期临床试验管理指导原则(试行)》起草说明 国家食品药品监督管理局 二O一一年十二月二日药物Ⅰ期临床试验管理指导原则(试行)第一章 总则 第一条 为加强药物Ⅰ期临床试验(以下简称I期试验)的管理,有效地保障受试者的权益与安全,提高Ⅰ期试验的研究质量与管理水平,根据《中华人民共和国药品管理法》、《药品注册管理办法》、《药物临床试验质量管理规范》等相关规定,参照国际通行规范,制定本指导原则。 第二条 本指导原则适用于Ⅰ期试验,旨在为Ⅰ期试验的组织管理和实施提供指导。人体生物利用度或生物等效性试验应参照本指导原则。第二章 职责要求 第三条 申办者应建立评价药物临床试验机构的程序和标准,选择、委托获得资格认定的I期试验研究室进行Ⅰ期试验。 第四条 申办者应建立质量保证体系,对I期试验的全过程进行监查和稽查,确保临床试验的质量,保障受试者的权益与安全。 第五条 申办者可以委托合同研究组织(CRO)执行I期试验中的某些工作和任务。委托前对合同研究组织的研究条件、能力、经验以及相应的质量管理体系进行评价。当合同研究组织接受了委托,则本指导原则中规定的由申办者履行的责任,合同研究组织应同样履行。申办者对临床试验的真实性及质量负最终责任。 第六条 Ⅰ期试验研究室负责Ⅰ期试验的实施。研究者应遵循临床试验相关法律法规、规范性文件和技术指导原则,执行临床试验方案,保护受试者的权益与安全,保证临床试验结果的真实可靠。 第七条 药物临床试验生物样本分析应在符合《药物临床试验生物样本分析实验室管理指南》(以下简称《实验室管理指南》)的实验室进行。从事药物临床试验生物样本分析的实验室均应接受药品监督管理部门的监督检查。 第八条 伦理委员会应针对Ⅰ期试验的特点,加强对受试者权益与安全的保护,重点关注:试验风险的管理与控制,试验方案设计和知情同意书的内容,研究团队的人员组成、资质、经验,受试者的来源、招募方式,实施过程中发生的意外情况等。
关于印发药物临床试验伦理审查工作指导原则的通知 国食药监注436号 2010年11月02日 发布 各省、自治区、直辖市食品药品监督管理局(药品监督管理局),总后卫生部药品监督管理局: 为加强药物临床试验的质量管理和受试者的保护,规范和指导伦理委员会的药物临床试验伦理审查工作,提高药物临床试验伦理审查工作质量,根据《药品注册管理办法》和《药物临床试验质量管理规范》的有关规定,国家局组织制定了《药物临床试验伦理审查工作指导原则》,现予印发。请你局指导本辖区内药物临床试验机构学习,参照执行。 附件:药物临床试验伦理审查工作指导原则起草说明 国家食品药品监督管理局 二○一○年十一月二日
实验数据中各变量的关系可表示为列表式,图示式和函数式。 列表式:将实验数据制成表格。它显示了各变量间的对应关系,反映出变量之间的变化规律。它是标绘曲线的基础。图示式:将实验数据绘制成曲线。它直观地反映出变量之间的关系。在报告与论文中几乎都能看到,而且为整理成数学模型(方程式)提供了必要的函数形式。 函数式:借助于数学方法将实验数据按一定函数形式整理成方程即数学模型。 熟悉相关和回归的定义,相关系数的定义,直线回归的最小二乘法。 熟悉药品质量标准分析方法验证中各项指标的定义和考察方法。 含量测定方法的评价 (效能指标—分析品质因数) 一般常用的分析效能评价指标包括:精密度、准确度、检测限、定量限、选择性、线性与范围、重现性、耐用性等;测定法的效能指标可评价分析测定方法,也可作为建立新的测定方法的实验研究依据。 1.精密度系指用该法测定同一匀质样品的一组测量值彼此符合的程度。它们越接近就越精密。在药物分析中,常用标准(偏)差(SD或S); 相对标准(偏)差(RSD),也称变异系数(CV),表示。 生物样品分析时,常用RSD表示精密度,并可细分为批内(或日内)精密度及批间(或日间)精密度。 批内精密度:是同一次测定的精密度。通常采用高、中、低三种浓度的同一样品各7-10份,每种浓度的样品按所拟定的分析方法操作,一次开机后,一一测定。计算每种浓度样品的SD值及RSD值。批内精密度也可视为日内精密度。所得RSD应争取达到5%以内,但不能超过10%。 批间精密度:是不同次测定的精密度。通常采用高、中、低三种浓度的同一样品,每种浓度配制7-10份,置冰箱冷冻。自配制样品之日开始,按所拟定的分析方法操作,每天取出一份测定,计算每种浓度样品的SD值及RSD值。批间精密度也可视为日间精密度。所得RSD应控制在15%以内。 2.准确度是指测得结果与真实值接近的程度,表示分析方法测量的正确性。 由于“真实值”无法准确知道,因此,通常采用回收率试验来表示。 制剂的含量测定时,采用在空白辅料中加入原料药对照品的方法作回收试验及计算RSD,还应作单独辅料的空白测定。每份均应自配制模拟制剂开始,要求至少测定高、中、低三个浓度,每个浓度测定三次,共提供9个数据进行评价。 回收率=(平均测定值M -空白值B)/ 加入量A×100% 回收率的RSD一般应为2%以内。 3.检测限(LOD)是指分析方法能够从背景信号中区分出药物时,所需样品中药物的最低浓度,无需定量测定。 LOD是一种限度检验效能指标,它既反映方法与仪器的灵敏度和噪音的大小,也表明样品经处理后空白(本底)值的高低。要根据采用的方法来确定检测限。当用仪器分析方法时,可用已知浓度的样品与空白试验对照,记录测得的被测药物信号强度S与噪音(或背景信号)强度N,以能达到S/N=2或S/N=3时的样品最低药浓为LOD;也可通过多次空白试验,求得其背景响应的标准差,将三倍空白标准差(即3δ空或3S空)作为检测限的估计值。为使计算得到的LOD值与实际测得的LOD值一致,可应用校正系数来校正,然后依之制备相应检测限浓度的样品,反复测试来确定LOD。如用非仪器分析方法时,即通过已知浓度的样品分析来确定可检出的最低水平作为检测限。 4.定量限 (LOQ)是指在保证具有一定可靠性(一定准确度和精密度)的前提下,分析方法能够测定出的样品中药物的最低浓度。 它反映了分析方法测定低药物浓度样品时具有的可靠性。它与上述的检测限的差别在于:定量限要定量测定某一药物在样品介质中的最低浓度,且定量限规定的最低浓度应该符合一定的精密度和准确度的要求。确定定量限的方法也因所用方法不同而异。当用非仪器分析方法时,与上述检测限的确定方法相同;如用仪器分析方法时,则往往将多次空白试验测得的背景响应的标准差(即空白标准差)乘以10,作为定量限的估计值,继之,再通过分析适当数量已知接近定量限或以定量限制备的样品来验证。 5.选择性是指在样品介质中有其他组分共存时该分析方法对供试物质准确而专属的测定能力。 它与专属性的含义稍有不同。专属性是指一种方法仅对一种分析成分产生唯一信号;选择性则可对多种化学成分产生不同响应,而主要成分的响应可与其它响应区分。 因此,选择性是指该法用于复杂样品分析时相互干扰程度的量度。 在药物分析中考察一个分析方法的选择性时,应着重考虑杂质、降解产物、相关化合物以及制剂辅料等其他组分是否对被测药物的测定有干扰。一般,通过添加上述物质的样品与未曾添加的样品所得分析结果进行比较而确定。 6.线性与范围 分析方法的线性是在给定范围内获取与样品中供试物浓度成正比的试验结果的能力。换句话说,就是供试物浓度的变化与试验结果(或测得的响应信号)成线性关系。 所谓线性范围是指利用一种方法取得精密度、准确度均符合要求的试验结果,而且成线性的供试物浓度的变化范围,其最大量与最小量之间的间隔,可用mg/L ~ mg/L、 ug/ml ~ ug/ml等表示。 线性与范围的确定可用作图法(响应值Y/浓度X)或计算回归方程(Y=a+bX)来研究建立。 测定样品时所有生物药物分析方法都必须同时作标准曲线。每次作标准曲线时,方法应与分析方法考核时完全一致。标准浓度应包括一定梯度的5-8个浓度(非线性者如免疫分析可适当增加),每个浓度只需测定一次(免疫分析可测定两次并取均值);标准曲线应覆盖样品可能的浓度范围,对于含量测定要求一般浓度上限为样品最高浓度的120%,下限为样品最低浓度的80% (但应高于LOQ);目前仍广泛采用相关系数(r)表示标准曲线的线性度、并控制r≥0.9900。对照品的LOQ必须包括在线性范围。 7.耐用性 是指利用相同的方法在各种正常实验条件下对同一样品进行分析所得结果的重现程度。 所谓各种正常实验条件,包括不同的实验室、不同的分析人员、不同的仪器、不同批号的试剂、不同的测试耗用时间、不同的分析温度、不同的测定日期等等。分析方法重现性的测定是通过在不同实验室由不同的实验者(操作和环境条件虽有差别但仍在规定的分析参数内)对同一样品的分别测试而获得的。 重现性 即是指在不同实验室中使用此种分析方法的精密度。是评价其保持不受参数微小变差影响的能力,并可作为正常使用的一个可靠性指标。 8. 与参比方法测得结果的相关程度的比较 由于生物样品中含有许多干扰测定的杂质,特别是与原型药物相似的代谢物常对药物的测定有影响。因此,除考察选择性外,有时还用参比方法对实际生物样品同时测定并进行比较。比较试验时,取若干份实际样品 (如病人服药后采取的血样),用一个已证明有相当专属性和可靠性的方法与新建立的方法同时进行测定,以参比方法测得的药浓为横坐标(X),以新建立方法测得的药浓为纵坐标(Y)作成散布图,并求出直线回归方程 (y=a+bx)及相关系数 (r)。r最大值为1,表示两法完全相关(结果完全吻合);r=0时,表示两法完全不相关。一般要求两法的相关系数r>0.95,而相关直线的斜率 应接近于1。 评价一种分析方法的效能,一般根据方法的使用对象区别。有以下四种情况: A.用于原料药中主要组分或制剂中有效组分含量测定的方法:除了检测限和定量限二项指标外,对精密度、准确度、选择性、线性与范围、耐用性等均应有所要求; B.用于原料药中杂质测定或制剂中降解产物测定的方法又可分为两种: ①用于含量测定;要求是:除检测限和精密度指标不必要求外,对准确度、选择性、线性与范围、定量限、耐用性等均应有所要求; ②用于限度检查。要求是:只对检测限、选择性和耐用性三项指标有所要求,其余均无需要求。 C.用于溶出度测定的方法及药物释放度测定的方法,只有精密度和耐用性有所要求,其余项目均不作要求。 D.用于生物样品中药物测定的方法,对精密度、准确度、检测限、选择性、可测线性范围、定量限、对生物样品的耐用性以及与参比方法测得结果的相关程度的比较等指标应有所要求。
[url=http://www.f-lab.cn/vivo-imaging/msot.html][b]小动物光声成像系统MSOT[/b][/url]是全球唯一能够提供[b]小动物全身光声成像[/b]能力的小动物[b]实时光声成像系统[/b],用于临床前小动物成像和临床前研究。小动物光声成像系统能够可帮助生物过程和药理物质作用在体内,在深部组织中高分辨率下实时观察。小动物光声成像系统是全球唯一[b]混合光声超声成像技术,OPUS成像[/b]技术的同类仪器,也是世界上第一个[b]交叉断层成像系统[/b],提供非平行的用户独立的图像质量,并且具有实时性,可以获得整个动物的横截面影像。[img=小动物光声成像系统]http://www.f-lab.cn/Upload/MOST-invision-imaging.JPG[/img][img=小动物光声成像系统]http://www.f-lab.cn/Upload/Hybrid-OPUS-IMAGING.jpg[/img]小动物光声成像系统:[url]http://www.f-lab.cn/vivo-imaging/msot.html[/url]
就对于,目前日系的汽车行业的标准来说,有些耐光照试验后需要评价褪色和变色,请问褪色和变色有什么区别呢?谢谢
药品的稳定性是指原料药及制剂保持其物理、化学、生物学和微生物学的性质,通过对原料药和制剂在不同条件(如温度、湿度、光线等)下稳定性的研究,掌握药品质量随时间变化的规律,为药品的生产、包装、贮存条件和有效期的确定提供依据,以确保临床用药的安全性和临床疗效。 稳定性研究是药品质量控制研究的主要内容之一,与药品质量研究和质量标准的建立紧密相关。稳定性研究具有阶段性特点,贯穿药品研究与开发全的过程,一般始于药品的临床前研究,在药品临床研究期间和上市后还应继续进行稳定性研究。 本文为一般性原则,具体的试验设计和评价应遵循具体问题具体分析的原则。 二、稳定性研究设计的考虑要素 稳定性研究的设计应根据不同的研究目的,结合原料药的理化性质、剂型的特点和具体的处方及工艺条件进行。 (一)样品的批次和规模 一般地,影响因素试验采用一批样品进行,加速试验和长期试验采用三批样品进行。 稳定性研究应采用一定规模生产的样品,以能够代表规模生产条件下的产品质量。原料药的合成工艺路线、方法、步骤应与生产规模一致;药物制剂的处方、制备工艺也应与生产规模一致。 稳定性研究中,原料药的供试品量应满足其制剂稳定性试验所要求的用量。口服固体制剂如片剂、胶囊应为10000个制剂单位左右。大体积包装的制剂(如静脉输液等)每批中试规模的数量至少应为各项试验所需总量的10倍。特殊品种、特殊剂型所需数量,视具体情况而定。 (二)包装及放置条件 稳定性试验要求在一定的温度、湿度、光照条件下进行,这些放置条件的设置应充分考虑到药品在贮存、运输及使用过程中可能遇到的环境因素。 原料药的加速试验和长期试验所用包装应采用模拟小包装,所用材料和封装条件应与大包装一致。药物制剂应在影响因素试验结果基础上选择合适的包装,在加速试验和长期试验中的包装应与拟上市包装一致。 稳定性研究中所用设备应能较好地对各项试验条件的要求的环境参数进行控制和监测。 (三)考察时间点 由于稳定性研究目的是考察药品质量随时间变化的规律,因此研究中一般需要设置多个时间点考察样品的质量变化。 考察时间点应基于对药品的理化性质的认识、稳定性趋势评价的要求而设置。如长期试验中,总体考察时间应涵盖所预期的有效期,中间取样点的设置要考虑药品的稳定性特点和剂型特点。对某些环境因素敏感的药品,应适当增加考察时间点。 (四)考察项目 稳定性研究的考察项目应选择在药品保存期间易于变化,并可能会影响到药品的质量、安全性和有效性的项目,以便客观、全面地反映药品的稳定性。根据药品特点和质量控制的要求,尽量选取能灵敏反映药品稳定性的指标。 一般地,考察项目可分为物理、化学、生物学和微生物学等几个方面。具体品种的考察项目设置应结合药品的特性进行。 (五)显著变化 稳定性研究中如样品发生了显著变化,则试验应中止。一般来说,原料药的“显著变化”应包括: 1、性状,如颜色、熔点、溶解度、比旋度超出标准规定,及晶型、水分等变化超出标准规定。 2、含量测定超出标准规定。 3、有关物质,如降解产物、异构体的变化等超出标准规定。 4、结晶水发生变化。 一般来说,药物制剂的“显著变化”包括: 1、含量测定中发生5%的变化;或者不能达到生物学或者免疫学检测过程的效价指标。 2、药品的任何一个降解产物超出标准规定。 3、性状、物理性质以及特殊制剂的功能性试验(如颜色、相分离、再混悬能力、结块、硬度、每揿给药剂量等)超出标准规定。 4、pH值超出标准规定; 5、制剂溶出度或释放度超出标准规定。 (六)分析方法 评价指标所采用的分析方法应经过充分的验证,能满足研究的要求,具有一定的专属性、准确性、灵敏度、重现性等。
求助!谁有药物分析实验室建设方案。现在系里正在搞实验室建设规划方案,本人打算申报一个药物分析实验室,需要有一些建设思路、规划方面的资料,还有所需设备仪器型号价格、开设实验项目啥的,哪位高手有的话能否帮忙发一份参考一下。十分感谢!wygsgdhr@126.com







 我要推广仪器
我要推广仪器
 下载APP
下载APP