凝胶电泳(英语:Gel electrophoresis)或称胶体电泳 是一大类技术,被科学工作者用于分离不同物理性质(如大小、形状、等电点等)的分子。凝胶电泳通常用于分析用途,但也可以作为制备技术,在采用某些方法(如质谱(MS)、聚合酶链式反应(PCR)、克隆技术、DNA测序或者免疫印迹)检测之前部分提纯分子。凝胶电泳仪 - 使用方法凝胶电泳被广泛用于分子生物学、遗传学和生物化学:1.大的DNA或者RNA分子通常利用琼脂糖凝胶电泳(agarose gel electrophoresis)分离,也可以使用聚丙烯酰胺凝胶电泳(PAGE)。2.蛋白质的凝胶电泳通常在加入十二烷基硫酸钠的聚丙烯酰胺凝胶中进行(SDS-PAGE),或者非变性凝胶电泳,或二维电泳。 SDS-PAGE 蛋白质凝胶电泳图。 3.毛细管电泳 4.酶谱法(zymography) 5.变性梯度胶凝电泳(Denaturing Gradient Gel Electrophoresis,DGGE) 琼脂糖和聚丙烯酰胺可以制成各种形状、大小和孔隙度。琼脂糖凝胶分离DNA片度大小范围较广,不同浓度琼脂糖凝胶可分离长度从200bp至近50kb的DNA片段。琼脂糖通常用水平装置在强度和方向恒定的电场下电泳。聚丙烯酰胺分离小片段DNA(5-500bp)效果较好,其分辩力极高,甚至相差1bp的DNA片段就能分开。聚丙烯酰胺凝胶电泳很快,可容纳相对大量的DNA,但制备和操作比琼脂糖凝胶困难。聚丙烯酰胺凝胶采用垂直装置进行电泳。目前,一般实验室多用琼脂糖水平平板凝胶电泳装置进行DNA电泳。 琼脂糖主要在DNA制备电泳中作为一种固体支持基质,其密度取决于琼脂糖的浓度。在电场中,在中性pH值下带负电荷的DNA向阳极迁移,其迁移速率由下列多种因素决定:1、 DNA的分子大小。度琼脂糖凝胶中的迁移速率与DNA分子量对数成反比,分子越大则所受阻力越大,也越难于在凝胶孔隙中蠕行,因而迁移得越慢。 2、 琼脂糖浓度 一个给定大小的线状DNA分子,其迁移速度在不同浓度的琼脂糖凝胶中各不相同。DNA电泳迁移率的对数与凝胶浓度成线性关系。凝胶浓度的选择取决于DNA分子的大小。分离小于0.5kb的DNA片段所需胶浓度是1.2-1.5%,分离大于10kb的DNA分子所需胶浓度为0.3-0.7%, DNA片段大小间于两者之间则所需胶浓度为0.8-1.0%。 3、 DNA分子的构象DNA分子处于不同构象时,它在电场中移动距离不仅和分子量有关,还和它本身构象有关。相同分子量的线状、开环和超螺旋DNA在琼脂糖凝胶中移动速度是不一样的,超螺旋DNA移动最快,而开环双链DNA移动最慢。如在电泳鉴定质粒纯度时发现凝胶上有数条DNA带难以确定是质粒DNA不同构象引起还是因为含有其他DNA引起时,可从琼脂糖凝胶上将DNA带逐个回收,用同一种限制性内切酶分别水解,然后电泳,如在凝胶上出现相同的DNA图谱,则为同一种DNA。
蛋白质的聚丙烯酰胺凝胶电泳 最广泛使用的不连续缓冲系统最早是由Ornstein(1964) 和Davis(1964) 设计的, 样品和浓缩胶中含 Tris-HCl(pH 6.8), 上下槽缓冲液含Tris-甘氨酸(pH 8.3), 分离胶中含Tris-HCl(pH 8.8)。系统中所有组分都含有0.1% 的 SDS(Laemmli, 1970)。样品和浓缩胶中的氯离子形成移动界面的先导边界而甘氨酸分子则组成尾随边界,在移动界面的两边界之间是一电导较低而电位滴度较陡的区域, 它推动样品中的蛋白质前移并在分离胶前沿积聚。此处pH值较高, 有利于甘氨酸的离子化,所形成的甘氨酸离子穿过堆集的蛋白质并紧随氯离子之后,沿分离胶泳动。从移动界面中解脱后,SDS-蛋白质复合物成一电位和pH值均匀的区带泳动穿过分离胶,并被筛分而依各自的大小得到分离。SDS与蛋白质结合后引起蛋白质构象的改变。SDS-蛋白质复合物的流体力学和光学性质表明,它们在水溶液中的形状,近似于雪茄烟形状的长椭园棒,不同蛋白质的SDS复合物的短轴长度都一样(约为18Å,即1.8nm),而长轴则随蛋白质分子量成正比地变化。这样的SDS-蛋白质复合物,在凝胶电泳中的迁移率,不再受蛋白质原有电荷和形状的影响,而只是椭园棒的长度也就是蛋白质分子量的函数。由于SDS和巯基乙醇的作用,蛋白质完全变性和解聚,解离成亚基或单个肽链,因此测定的结果只是亚基或单条肽链的分子量。 SDS聚丙烯酰胺凝胶的有效分离笵围取决于用于灌胶的聚丙烯酰胺的浓度和交联度。在没有交联剂的情况下聚合的丙烯酰胺形成毫无价值的粘稠溶液,而经双丙烯酰胺交联后凝胶的刚性和抗张强度都有所增加,并形成SDS蛋白质复合物必须通过的小孔。这些小孔的孔径随 “双丙烯酰胺~丙烯酰胺” 比率的增加而变小,比率接近 1:20 时孔径达到最小值。SDS聚丙烯酰胺凝胶大多按“双丙烯酰胺~丙烯酰胺”为1:29 配制,试验表明它能分离大小相差只有3% 的蛋白质。 凝胶的筛分特性取决于它的孔径,而孔径又是灌胶时所用丙烯酰胺和双丙烯酰胺绝对浓度的函数。用5~15%的丙烯酰胺所灌制凝胶的线性分离范围如下表:
奶粉蛋白质中乳清蛋白含量的测定 十二烷基硫酸钠-毛细管凝胶电泳法该标准还未正式发布
1.原理等电聚焦凝胶电泳是依据蛋白质分子的静电荷或等电点进行分离的技术,等电聚焦中,蛋白质分子在含有载体两性电解质形成的一个连续而稳定的线性pH梯度中电泳。载体两性电解质是脂肪族多氨基多羧酸,在电场中形成正极为酸性,负极为碱性的连续的pH梯度。蛋白质分子在偏离其等电点的pH条件下带有电荷,因此可以在电场中移动;当蛋白质迁移至其等电点位置时,其静电荷数为零,在电场中不再移动,据此将蛋白质分离。 等电聚焦中,只有在凝胶两端给以高电压时,才能获得较好的蛋白质条带分辨率,这就需要非常有效的凝胶冷却系统(否则会导致烧胶),即凝胶同期周围液体之间的热传递效率要高。由于平板胶热传递能力高,并可方便的同时比较多种蛋白质样品,所以平板胶用在等电聚焦上的居多。由于等电聚焦对蛋白质的电荷差异非常敏感,若要好的重复性,制备蛋白样品时一定要小心,要避免任何对蛋白质化学组成和结构的修饰。另外,蛋白质-脂类、蛋白质-蛋白质相互作用可引起电荷改变,进而导致等电点迁移或纹理现象。除非特殊需要研究蛋白质-蛋白质相互作用或者必须保持蛋白质的生物学功能,等电聚焦通常在含有尿素的变性凝胶系统中进行。使用非离子去垢剂也可以提高分辨率。 2.主要仪器、试剂仪器:微型电泳系统、电源、注射器、固定和染色用容器 试剂:丙稀酰胺、双-丙稀酰胺、载体两性电解质、尿素、过硫酸胺、TEMED、TritonX-100、2-巯基乙醇、溴酚蓝、磷酸、氢氧化钠、氯化钾、三氯乙酸、考马斯亮蓝、甲醇、乙酸。 储存液:1)30%(w/v)丙稀酰胺,1%(w/v)双-丙稀酰胺;2)20%Triton X100;3)10%三氯乙酸;4)1%三氯乙酸;5)1%溴酚蓝;6)考马斯亮蓝染色液;7)考马斯亮蓝脱色液;
[b]凝胶水平电泳仪[/b]和[b]进口水平电泳仪[/b]非常经济的低缓冲量,非常适合超大规模样品检测或高分辨率分离实验的[b]水平电泳仪,[url=http://www.f-lab.cn/electrophoresis/me-10.html]水平凝胶电泳仪[/url][/b][url=http://www.f-lab.cn/electrophoresis/me-10.html]FP-ME-10-20[/url]专业为克隆或PCR实验而设计,适合大规模的凝胶电泳实验和超高分辨率的分离,也适合样品转移到膜层上进行进一步分析。[b]水平凝胶电泳仪[/b]特色具有三种凝胶盘尺寸:200x200mm,200x250mm和200x100mm兼容多通道[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff][url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液器[/color][/url][/color][/url]的凝胶卡可一次性使用40个样品,单个凝胶可加载440个样品电泳液冲洗消耗少兼容多道[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff][url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液器[/color][/url][/color][/url]单个铸造电泳槽安全可靠方便样品加载电泳指示功能凝胶灌胶无需夹具,弹簧[b]水平凝胶电泳仪参数[/b]体积:W395xL230xH90mm凝胶卡尺寸: W200xL200mm, W200xL100mm 最大样品:200@W200xL200mm, 450@W200xL100mm 缓冲液容积:1200ml建造:注塑铸造,耐用防漏电极:99.99%铂金电极,可更换而抗腐蚀更多凝胶电泳仪请浏览官网:[url]http://www.f-lab.cn/electrophoresis.html[/url]
凝胶电泳仪器的发展 电泳系统虽然只是作为生化分离分析所必需的常规仪器,但它的进展与其他大型仪器设备一样,同样是非常迅速的。从1809年的第一次电泳实验所用的雏型装置到1946年第一台商品自由移界电泳系统问世经历了一个多世纪。但此后的50多年电泳仪器的发展却极其迅猛,特别是电泳介质由流动相改为凝胶后,各种各样的凝胶电泳装置便层出不穷以适应各种研究工作和生产实践的需要。
由于实验需要现准备购置一台二维凝胶电泳仪,价格适中最好,希望各位大侠帮忙推荐!先谢了![em23]
求助于高手们: 我要用毛细管凝胶电泳检测乳制品中的蛋白质,请问我的样品和电泳缓冲溶液用什么啊?
Difference Gel Electrophoresis (DIGE)即差异凝胶电泳是目前2D领域相当热门的一项技术,可以说它的出现消除了传统双向的一些弊端,使得好多实验室对双向重拾信心http://img.dxycdn.com/images_new/smiles/smile_tongue.gif 先简要介绍一下DIGE技术的历史和路线。DIGE技术最早在1997年由Jon Minden实验室提出,后来Amresham成为此项技术的主要推动者,其技术路线是将样品在电泳前标记Cy2、Cy3 和Cy5 这3 种荧光染料(其中Cy2 作为用Cy3、Cy5 标记的蛋白质内标, 用以比较数据库中各个胶之间的蛋白质的量的差异),然后将标记后的3 种样品混合, 同时在一块胶上进行电泳。所得到的2D 胶图像可使用3 种不同的激发P发射过滤器得到不同颜色荧光信号,根据这些信号的比例来判断样品之间蛋白质的差异,这样可避免在匹配时出现的误差,进行定量分析时也不依赖于胶与胶之间的重复性。用于标记的荧光基团在化学结构上相似,分子量也基本相同,都带有正电荷,所以在与赖氨酸残基反应时,保证了所有的样品可以移至相同的位置。该方法的灵敏度可与银染和SYPRO Ruby 相媲美,可以检测到100~200pg 的蛋白质,线形动态范围在5个数量级左右。该方法也提高了定量上的准确性。由于实验方法本身造成的蛋白质斑点强度的差异,比如样品在进入胶条时会有一些样品损失,但在同一块差异凝胶电泳胶上就不会出现这样的差异。DIGE 可在同一次检测中通过分析单一胶上的相互覆盖的信息来得到蛋白质的信息,可以同时在一块胶上在相同电泳条件下分离2~3 个样品,大大减少了一个实验需要的胶的总数。虽然从理论上来说,该方法非常诱人, 但DIGE 本身还有很多技术上的问题。主要的问题在于:1) 只有当约1 %~2 %的蛋白质的赖氨酸残基在荧光标记时修饰,才可以维持被标记的蛋白在电泳时的溶解性;2) 在DIGE 染色中丢失的蛋白质基本确定为样品中的低丰度蛋白; 3) 虽然经染色后在电荷上未发生变化,被染料标记的蛋白质比未标记的蛋白质点向大分子方向有轻微迁移,主要是由于荧光团的加入(500Da左右)使得在标记的和未标记蛋白之间产生95%的误差, 之后在质谱分析时产生更多的复杂问题。4) 另一个值得注意的影响灵敏度的问题是荧光物质在激发P发射过程中会出现信号的相互干扰,引发Cy3 的激发波长有时可引起Cy5 标记的蛋白发射。(以上内容部分摘自国外医学卫生学分册2004年第31卷第1期姜楠文章)
各位大哥,电泳仪怎么样维护比较好(主要做琼脂糖凝胶电泳的),还有一些注意事项的?比如……
根据电泳仪原理、电泳仪功能、电泳仪的使用方法、电泳仪的用途不同可以为:琼脂糖凝胶电泳、毛细管电泳、凝胶电泳、聚丙烯酰胺凝胶电泳、醋酸纤维薄膜电泳、高效毛细管电泳、琼脂糖电泳、SDS-PAGE凝胶电泳、蛋白质电泳、血清蛋白电泳、dna电泳、血红蛋白电泳、蛋白质双向电泳、免疫电泳、等电聚焦电泳、单细胞凝胶电泳、蛋白质凝胶电泳、质粒电泳、对流免疫电泳、变性电泳等。电泳仪分类:1、毛细管电泳仪:其主要部件有0~30kV可调稳压稳流电源,内径小于100μm(常用50~75μm)、长度一般为30~100cm的弹性石英毛细管、电极槽、检测器和进样装置。检测器有紫外/可见分光检测器、激光诱导荧光检测器和电化学检测器,前者最为常用。进样方法有电动法(电迁移)、压力法(正压力、负压力)和虹吸法。成套仪器还配有自动冲洗、自动进样、温度控制、数据采集和处理等部件。 2、常规电泳仪:其组成部件为可调稳压稳流电源,垂直电泳槽,水平电泳槽,电极连接线,支持体【非凝胶性支持体区带电泳(支持体有:①淀粉②纤维素粉③玻璃粉,硅胶等) ;凝胶支持体区带电泳支持体有:①淀粉液②聚丙烯酰胺凝胶③琼脂(糖)凝胶】;陶瓷板,抽水泵,输水管,冰水曹等部件组成。3、其他电泳仪:Tiselius或微量电泳、显微电泳、等电点聚焦电泳技术、等速电泳技术、密度梯度电泳等。是一种非支持体的电泳仪也称为自由电泳法的发展并不迅速,因为其电泳仪构造复杂、体积庞大,操作要求严格,价格昂贵等很少使用。毛细管电泳仪、凝胶电泳仪、垂直电泳仪、微电泳仪、高压电泳仪、水平电泳仪、高效毛细管电泳仪、双向电泳仪、bio rad 电泳仪、脉冲场电泳仪、伯乐电泳仪、北京六一电泳仪上海巴玖均可提供!
第一节 概 述一. DNA的限制性内切酶酶切分析限制性内切酶能特异地结合于一段被称为限制性酶识别序列的DNA序列之内或其附近的特异位点上,并切割双链DNA。它可分为三类:Ⅰ类和Ⅲ类酶在同一蛋白质分子中兼有切割和修饰(甲基化)作用且依赖于ATP的存在。Ⅰ类酶结合于识别位点并随机的切割识别位点不远处的DNA,而Ⅲ类酶在识别位点上切割DNA分子,然后从底物上解离。Ⅱ类由两种酶组成: 一种为限制性内切核酸酶(限制酶),它切割某一特异的核苷酸序列; 另一种为独立的甲基化酶,它修饰同一识别序列。Ⅱ类中的限制性内切酶在分子克隆中得到了广泛应用,它们是重组DNA的基础。绝大多数Ⅱ类限制酶识别长度为4至6个核苷酸的回文对称特异核苷酸序列(如EcoRⅠ识别六个核苷酸序列:5'- G↓AATTC-3'),有少数酶识别更长的序列或简并序列。Ⅱ类酶切割位点在识别序列中,有的在对称轴处切割,产生平末端的DNA片段(如SmaⅠ:5'-CCC↓GGG-3');有的切割位点在对称轴一侧,产生带有单链突出末端的DNA片段称粘性未端, 如EcoRⅠ切割识别序列后产生两个互补的粘性末端。5'…G↓AATTC…3' →5'… G AATTC…3'3'…CTTAA↑G …5' →3'… CTTAA G…5'DNA纯度、缓冲液、温度条件及限制性内切酶本身都会影响限制性内切酶的活性。大部分限制性内切酶不受RNA或单链DNA的影响。当微量的污染物进入限制性内切酶贮存液中时,会影响其进一步使用,因此在吸取限制性内切酶时,每次都要用新的吸管头。如果采用两种限制性内切酶,必须要注意分别提供各自的最适盐浓度。若两者可用同一缓冲液,则可同时水解。若需要不同的盐浓度,则低盐浓度的限制性内切酶必须首先使用,随后调节盐浓度,再用高盐浓度的限制性内切酶水解。也可在第一个酶切反应完成后,用等体积酚/氯仿抽提,加0.1倍体积3mol/L NaAc和2倍体积无水乙醇,混匀后置-70℃低温冰箱30分钟,离心、干燥并重新溶于缓冲液后进行第二个酶切反应。DNA限制性内切酶酶切图谱又称DNA的物理图谱,它由一系列位置确定的多种限制性内切酶酶切位点组成,以直线或环状图式表示。在DNA序列分析、基因组的功能图谱绘制、DNA的无性繁殖、基因文库的构建等工作中,建立限制性内切酶图谱都是不可缺少的环节,近年来发展起来的RFLP(限制性片段长度多态性)技术更是建立在它的基础上。构建DNA限制性内切酶图谱有许多方法。通常结合使用多种限制性内切酶,通过综合分析多种酶单切及不同组合的多种酶同时切所得到的限制性片段大小来确定各种酶的酶切位点及其相对位置。酶切图谱的使用价值依赖于它的准确性和精确程度。在酶切图谱制作过程中,为了获得条带清晰的电泳图谱,一般DNA用量约为0.5-1μg。限制性内切酶的酶解反应最适条件各不相同,各种酶有其相应的酶切缓冲液和最适反应温度(大多数为37℃)。对质粒DNA酶切反应而言, 限制性内切酶用量可按标准体系1μg DNA加1单位酶,消化1-2小时。但要完全酶解则必须增加酶的用量,一般增加2-3倍,甚至更多,反应时间也要适当延长。二. 凝胶电泳琼脂糖或聚丙烯酰胺凝胶电泳是分离鉴定和纯化DNA片段的标准方法。该技术操作简便快速,可以分辨用其它方法(如密度梯度离心法)所无法分离的DNA片段。当用低浓度的荧光嵌入染料溴化乙啶(Ethidium bromide, EB)染色,在紫外光下至少可以检出1-10ng的DNA条带,从而可以确定DNA片段在凝胶中的位置。此外,还可以从电泳后的凝胶中回收特定的DNA条带,用于以后的克隆操作。琼脂糖和聚丙烯酰胺可以制成各种形状、大小和孔隙度。琼脂糖凝胶分离DNA片度大小范围较广,不同浓度琼脂糖凝胶可分离长度从200bp至近50kb的DNA片段。琼脂糖通常用水平装置在强度和方向恒定的电场下电泳。聚丙烯酰胺分离小片段DNA(5-500bp)效果较好,其分辩力极高,甚至相差1bp的DNA片段就能分开。聚丙烯酰胺凝胶电泳很快,可容纳相对大量的DNA,但制备和操作比琼脂糖凝胶困难。聚丙烯酰胺凝胶采用垂直装置进行电泳。目前,一般实验室多用琼脂糖水平平板凝胶电泳装置进行DNA电泳。琼脂糖主要在DNA制备电泳中作为一种固体支持基质,其密度取决于琼脂糖的浓度。在电场中,在中性pH值下带负电荷的DNA向阳极迁移,其迁移速率由下列多种因素决定:1、DNA的分子大小:线状双链DNA分子在一定浓度琼脂糖凝胶中的迁移速率与DNA分子量对数成反比,分子越大则所受阻力越大,也越难于在凝胶孔隙中蠕行,因而迁移得越慢。2、琼脂糖浓度一个给定大小的线状DNA分子,其迁移速度在不同浓度的琼脂糖凝胶中各不相同。DNA电泳迁移率的对数与凝胶浓度成线性关系。凝胶浓度的选择取决于DNA分子的大小。分离小于0.5kb的DNA片段所需胶浓度是1.2-1.5%,分离大于10kb的DNA分子所需胶浓度为0.3-0.7%, DNA片段大小间于两者之间则所需胶浓度为0.8-1.0%。
Q:SDS-PAGE电泳的基本原理?A:SDS-聚丙烯酰胺凝胶电泳,是在聚丙烯酰胺凝胶系统中引进SDS(十二烷基硫酸钠),SDS会与变性的多肽,并使蛋白带负电荷,由于多肽结合SDS的量几乎总是与多肽的分子量成正比而与其序列无关,因此SDS多肽复合物在丙稀酰胺凝胶电泳中的迁移率只与多肽的大小有关,在达到饱和的状态下,每克多肽可与1.4g去污剂结合。当分子量在15KD到200KD之间时,蛋白质的迁移率和分子量的对数呈线性关系,符合下式:logMW=K-bX,式中:MW为分子量,X为迁移率,k、b均为常数,若将已知分子量的标准蛋白质的迁移率对分子量对数作图,可获得一条标准曲线,未知蛋白质在相同条件下进行电泳,根据它的电泳迁移率即可在标准曲线上求得分子量。
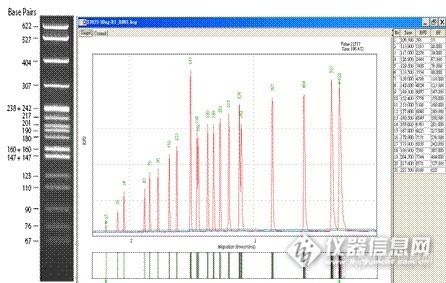
毛细管电泳技术(Capillary Electrophoresis, CE)又称高效毛细管电泳(HPCE)或毛细管分离法(CESM),是一类以毛细管为分离通道、以高压直流电场为驱动力,根据样品中各组分之间迁移速度和分配行为上的差异而实现分离的一类液相分离技术,迅速发展于80年代中后期,它实际上包含电泳技术和色谱技术及其交叉内容,是分析科学中继高效液相色谱之后的又一重大进展。http://ng1.17img.cn/bbsfiles/images/2013/08/201308081111_456810_2646159_3.jpg1987年,Cohen发表了毛细管凝胶电泳的工作。当电泳从凝胶板上移到毛细管中以后,发生了奇迹般的变化:分析灵敏度提高到能检测一个碱基的变化,分离效率达百万理论塔板数;分析片段能大能小,小到分辨单个核苷酸的序列,大到分离Mb的DNA;分析时间由原来的以小时计算缩减到以分、秒计算。CE可以说是经典电泳技术与现代微柱分离技术完美结合的产物。 一.毛细管凝胶电泳的原理 不同分子所带电荷性质、多少不同,形状、大小各异。一定电解质及PH的缓冲液或其它溶液内,受电场作用,样本中各组分按一定速度迁移,从而形成电泳。电泳迁移速度(v)可用下式表示:v=uE其中E为电场强度(E=V/L,V为电压,L为毛细管总长度)。u为电泳淌度。毛细管凝胶电泳是将板上的凝胶移到毛细管中作支持物进行的电泳。凝胶具有多孔性,起类似分子筛的作用, 溶质按分子大小逐一分离。凝胶粘度大, 能减少溶质的扩散, 所得峰形尖锐, 能达到CE中最高的柱效。电流通过导体时产生焦耳热。传统平板凝胶电泳的最大局限性在于其无法克服两端高电压带来的焦耳热所产生的负面影响。焦耳热可使筛分介质内部出现温度、粘度及分离速度的不均一,影响迁移、降低效率、使区带变宽。由于这种负面影响与电场强度成正比,所以极大地限制了高电压的引入。也难以提高电泳速度。毛细管电泳使样品在一根极细的柱子中进行分离。细柱可减小电流,使焦耳热的产生减少;同时又增大了散热面积,提高散热效率,大大降低了管中心与管壁间的温差,减少了柱子径向上的各种梯度差,保证了高效分离。因此可以加大电场强度,达到100~200V/cm,全面提高分离质量。在进行分析时将毛细管内充满了凝胶,毛细管两端通高压电,使凝胶内带电分子移到毛细管相反电荷的一端。因为不同分子的大小对电荷比不同,就以不同的速率在管中移动,达到毛细管终点也有快有慢。毛细管电泳即依此探测、分离不同分子。 二.毛细管凝胶电泳的特点 1.所需样品量少、仪器简单、操作简便。2.分析速度快,分离效率高,分辨率高,灵敏度高。3.无需核酸染料,安全无毒。4.无需制胶,省时省力。5.无需照胶,杜绝人工分析结果误差。6.自动出结果,包括片段大小和样品浓度,软件可输出电泳峰图、凝胶电泳图、DNA片段碱基差异分析、相对定量分析。http://ng1.17img.cn/bbsfiles/images/2013/08/201308081111_456811_2646159_3.jpg三.毛细管凝胶电泳的应用1.PCR条件的优化及多重PCR的检测2.高分辨率的检出,相差1-4bp的DNA片段的差异,及相同长度不同序列的差异3.动态检测酶切体系的进程4.评估基因组DNA质量高低5.HLA分型6.STR分析7.质粒的纯度分析8.DNA/DNA杂交9.DNA/蛋白互作
[size=4][b][size=5][font=黑体][/font][/size] [/b][/size][b]SDS聚丙烯酰胺凝胶电泳原理[/b] 采用十二烷基硫酸钠-聚丙稀酰胺凝胶电泳(SDS-PAGE,polyacrylamide gel electrophoresis)方法可对蛋白质的组分进行分离,并可精确测得蛋白质的分子量。常用的方法为SDS-PAGE不连续系统。基本原理:聚丙稀酰胺是由丙稀酰胺(acrylamide)和N,N’-亚甲基双丙稀酰胺(N,N’-methylene bis acrylamide)经共聚合而成。此聚合过程是由四甲基乙二胺(tetramethylethylenediamine,TEMED)和过硫酸胺(ammonium persulfate,AP)激发的。被激活的单体和未被激活的单体开始了多聚链的延伸,正在延伸的多聚链也可以随机地接上双丙稀酰胺,使多聚链交叉互连成为网状立体结构,最终多聚链聚合成凝胶状。[b]丙烯酰胺是一种白色晶体化学物质,是生产聚丙烯酰胺的原料。聚丙烯酰胺主要用于水的净化处理、纸浆的加工及管道的内涂层等。淀粉类食品在高温( 120℃)烹调下容易产生丙烯酰胺。[/b] [b]研究表明,人体可通过消化道、呼吸道、皮肤黏膜等多种途径接触丙烯酰胺,饮水是其中的一条重要接触途径。[/b] [b]丙烯酰胺进入体内又可通过多种途径被人体吸收,其中经消化道吸收最快。进入人体内的丙烯酰胺约90%被代谢,仅少量以原形经尿液排出。丙烯酰胺进入体内后,会在体内与DNA上的鸟嘌呤结合形成加合物,导致遗传物质损伤和基因突变。[/b] [b]对接触丙烯酰胺的职业人群和偶然暴露于丙烯酰胺人群的调查表明,丙烯酰胺具有神经毒性作用,但目前还没有充足的证据表明通过食物摄入丙烯酰胺与人类某种肿瘤的发生有明显关系。[/b][b][/b] [b]丙烯酰胺简介[/b][b]丙烯酰胺是一种有机化合物,别名AM;纯品为白色结晶固体,易溶于水、甲醇、乙醇、丙醇,稍溶于乙酸乙酯、氯仿,微溶于苯,在酸碱环境中可水解成丙烯酸。职业性接触主要见于丙烯酰胺生产和树脂、黏合剂等的合成,在地下建筑、改良土壤、油漆、造纸及服装加工等行业也有接触机会。日常生活中,丙烯酰胺可见于吸烟、经高温加工处理的淀粉食品及饮用水中。[/b][毒性] 丙烯酰胺属中等毒类,对眼睛和皮肤有一定的刺激作用,可经皮肤、呼吸道和消化道吸收,在体内有蓄积作用,主要影响神经系统,急性中毒十分罕见。密切大量接触可出现亚急性中毒,中毒者表现为嗜睡、小脑功能障碍以及感觉运动型多发性周围神经病。长期低浓度接触可引起慢性中毒,中毒者出现头痛、头晕、疲劳、嗜睡、手指刺痛、麻木感,还可伴有两手掌发红、脱屑,手掌、足心多汗,进一步发展可出现四肢无力、肌肉疼痛以及小脑功能障碍等。丙烯酰胺慢性毒性作用最引人关注的是它的致癌性。丙烯酰胺具有致突变作用,可引起哺乳动物体细胞和生殖细胞的基因突变和染色体异常。动物试验研究发现,丙烯酰胺可致大鼠多种器官肿瘤,如乳腺、甲状腺、睾丸、肾上腺、中枢神经、口腔、子宫、脑下垂体肿瘤等。但目前还没有充足的人群流行病学证据表明,食物摄入丙烯酰胺与人类某种肿瘤的发生有明显相关性。国际癌症研究机构(IARC)对其致癌性进行了评价,将丙烯酰胺列为2类致癌物(2A),即人类可能致癌物。其主要依据为,丙烯酰胺在动物和人体均可代谢转化为致癌活性代谢产物环氧丙酰胺。[预防]⒈职业性接触者要通过改革工艺、采取工程技术措施等手段,降低工作场所空气中丙烯酰胺的浓度;同时通过加强个人防护,如戴口罩、手套,穿防护服和鞋等,以防止或减少丙烯酰胺进入体内。⒉日常生活中尽量避免过度烹饪食品,如温度过高或加热时间太长。提倡平衡膳食,减少油炸和高脂肪食品的摄入,多吃水果和蔬菜,不要吸烟。⒊由于煎炸食品是我国居民常吃的食物,国家应加强膳食中丙烯酰胺的监测与控制,开展我国人群丙烯酰胺的暴露评估,并研究探索减少加工食品中丙烯酰胺含量的方法 [b]N.N-亚甲基双丙烯酰胺,别名MBA,双叫N.N-甲叉双丙烯酰胺,次甲基双丙烯酰胺,N.N-甲撑双丙烯酰胺。是一种白色晶体粉末,无味,吸湿性极小。遇高温或强光则自交联,微溶于水、乙醇。[/b][b]丙烯酰胺单体和交联剂N1 N′-亚甲基双丙烯酰胺在催化剂的作用下聚合成含有酰胺基侧链的脂肪族长链。相邻的两个链通过亚甲基桥交联起来就形成三维网状结构的聚丙烯酰胺凝胶。[/b][b]N, N -亚甲基双丙烯酰胺又名甲撑双丙烯酰胺 , 英文缩写名 MBA, 为白色或浅黄色粉末状结晶 , 毒性低 , 对皮肤无刺激 , 无神经毒性 , 溶于水及乙醇、丙酮等有机溶剂。在它的结构中具有两个相同且非常活泼的反应性官能团 , 可作为交联剂 ,能将线性高分子迅速转变为体型高分子 , 制备吸水性聚合物 , 还可与各种离子型单体发生聚合反应 ,使其在石油开采以及医药、水处理等行业具有广泛用途。 [/b][b]产品简介: [/b][b]? TEMED即N,N,N‘,N’-Tetramethylethylenediamine,中文名为N,N,N‘,N’-四甲基二乙胺。分子式为(CH3)2NCH2CH2N(CH3)2, 分子量为116.20。 [/b][b]? 进口分装,用于配制PAGE胶等。TEMED通过催化过硫酸铵形成自由基而加速丙烯酰胺与双丙烯酰胺的聚合。[/b][b]? 加入加速剂TEMED后聚合马上开始,应立即将凝胶混匀,迅速灌胶。[/b][b]保存条件: 4℃保存。 [/b][b]注意事项: [/b][b]?易燃,有腐蚀性,请注意防护。 [/b][b]?为了您的安全和健康,请穿实验服并戴一次性手套操作。[/b][b]过硫酸铵分子式: (NH4)2S2O8 分子量: 228.20[/b][b]性状:过硫酸铵是一种白色、无味晶体,常作强氧化剂使用,也可用作单体聚合引发剂。它几乎不吸潮,由于能达到很高的纯度而具有特别好的稳定性,便于储存。另外,它还具有使用方便、安全等优点。[/b][b]储存及使用注意事项:[/b] [b]过硫酸铵属于非易燃品,但由于能释放氧而有助燃作用,因此必须在一定条件下储存。首先必须存放在干燥、密闭的容器中,其次应避免阳光直射、热源、潮湿等不利因素。另外,一些杂质如脏物、铁锈、少量金属以及还原剂可能引起过硫酸铵的分解,在存放和使用过程中也必须注意。由于潮湿的过硫酸铵粉末及其水溶液有漂白和轻微的腐蚀作用,因此使用过程中应避免眼睛、皮肤和衣物直接与其接触。[/b][b]过硫酸铵的应用:过硫酸铵提供驱动丙烯酰胺和双丙烯酰胺聚合所必需的自由基。须新鲜配制[/b]。 [b]过硫酸铵是乳胶或丙烯酸单体聚合液、醋酸乙烯、氯乙烯等产品的引发剂,同时也是苯乙烯、丙烯腈、丁二烯等胶体发生共聚作用的引发剂。 [/b][b]过硫酸铵-TEMED(四甲基乙二胺)系统:在Acr 和Bis的溶液中放入这个催化系统后,过硫酸铵[(NH4)2S2O8]产生出游离氧原子使单体成为具有游离基的状态,从而发生聚合作用。聚合的初速度和过硫酸铵浓度的平方根成正比。这种催化系统需要在碱性条件下进行。例如,在pH 8.8条件下7%的丙烯酰胺溶液30分钟就能聚合完毕;在 pH 4.3时聚合很慢,要90分钟才能完成。温度与聚合的快慢成正比。通常在室温下就很快聚合,温度升高聚合更快。如将混合后的凝胶溶液放在近0℃的地方,就能延缓聚合。一般来讲,温度过低,有氧分子或不纯物质存在时都能延缓凝胶的聚合。为了防止溶液中气泡含有氧分子而妨碍聚合,在聚合前须将溶液分别抽气,然后再混合。[/b][b]十二烷基硫酸钠 SDS[/b][b]不连续系统由上层浓缩胶和下层的分离胶组成。浓缩胶(pH6.7,孔径大)主要作用是使样品浓缩,使样品在未进入分离胶前,被浓缩成很窄的条带,从而提高分离效果。分离胶(pH8.9,孔径小)通过分子筛效应和电荷效应,把样品中的各组分按分子量和电荷的大小而分开。 [/b]如果要利用凝胶电泳测定某一蛋白质的分子量就必须将电荷效应去掉或减少到可以忽略不计的程度,使蛋白质泳动率的大小完全取决于分子量。如何去除电荷效应呢?现常用的是十二烷基硫酸钠(SDS)。SDS是一种阴离子去污剂。在电泳体系中加入一定浓度的SDS,SDS以一定的比例和蛋白质分子结合成复合物,使蛋白质分子带负电荷,这种负电荷远远超过了蛋白质分子原有的电荷,从而减低或消除了各种蛋白质分子天然电荷的差异。[b]是阴离子型表面活性剂,它能按一定比例与蛋白质分子结合成带负电荷的复合物,再与PAGE技术结合,则谱带差异更加明显、清晰,并可测定蛋白质分子量。 [/b][b]在有去污剂十二烷基硫酸钠存在下的聚丙烯酰胺凝胶电泳。SDS-PAGE只是按照分子大小分离的,而不是根据分子所带的电荷和大小分离的。[/b][b]SDS带有大量负电荷,当其与蛋白质结合时,所带的负电荷大大超过了蛋白质原有的负电荷,因而消除或掩盖了不同种类蛋白质间原有电荷的差异,使蛋白质均带有相同密度的负电荷,因而可利用Mr差异将各种蛋白质分开。[/b][b]甘氨酸[/b][b]最广泛使用的不连续缓冲系统最早是由Ornstein(1964) 和Davis(1964) 设计的, 样品和浓缩胶中含 Tris-HCl(pH 6.8), 上下槽缓冲液含Tris-甘氨酸(pH 8.3), 分离胶中含Tris-HCl(pH 8.8)。系统中所有组分都含有0.1% 的 SDS(Laemmli, 1970)。样品和浓缩胶中的氯离子形成移动界面的先导边界而甘氨酸分子则组成尾随边界,在移动界面的两边界之间是一电导较低而电位滴度较陡的区域, 它推动样品中的蛋白质前移并在分离胶前沿积聚。此处pH值较高,有利于甘氨酸的离子化,所形成的甘氨酸离子穿过堆集的蛋白质并紧随氯离子之后,沿分离胶泳动。从移动界面中解脱后,SDS-蛋白质复合物成一电位和pH值均匀的区带泳动穿过分离胶,并被筛分而依各自的大小得到分离。[/b][b]浓缩效应:凝胶由两种不同的凝胶层组成。上层为浓缩胶,下层为分离胶。浓缩胶为大孔胶,缓冲液pH6.7,分离胶为小孔胶,缓冲液pH8.9。在上下电泳槽内充以Tris—甘氨酸缓冲液(pH8.3),这样便形成了凝胶孔径和缓冲液pH值的不连续性。在浓缩胶中 HCl几乎全部解离为Cl-,但只有极少部分甘氨酸解离为H2NCH2COO-。蛋白质的等电点一般在pH5左右,在此条件下其解离度在HCl和甘氨酸之间。当电泳系统通电后,这3种离子同向阳极移动。其有效泳动率依次为Cl->蛋白质>H2NCH2COO-,故C1-称为快离子,而H2NCH2COO- 称为慢离子。电泳开始后,快离子在前,在它后面形成离子浓度低的区域即低电导区。电导与电压梯度成反比,所以低电导区有较高的电压梯度。这种高电压梯度使蛋白质和慢离子在快离子后面加速移动。在快离子和慢离子之间形成—个稳定而不断向阳极移动的界面。由于蛋白质的有效移动率恰好介于快慢离子之间,因此蛋白质离子就集聚在快慢离子之间被浓缩成—条狭窄带。这种浓缩效应可使蛋白质浓缩数百倍。 [/b]
请问:国产和进口变性梯度凝胶电泳一般都在什么价位。比较好的品牌有那几个,谢谢!
本实验讲义是在接受了去年实验的成功经验和失败的教训的基础上编写的。实验的名字叫“分子生物学技术综合实验”,原来是想把它设计成一个彼此连贯、前后呼应的系列实验。但由于经费和条件的限制,还是放弃了这个念头。 实验分成两个部分:前一部分是DNA的操作,主要是DNA片段的亚克隆的方法;后一部分是蛋白的操作,主要是凝胶电泳和免疫杂交。两个部分的分量大致相当,但后一部分做起来对学生的收获可能更大些,因为很少有本科生能一个人原原本本地亲手做一做免疫印迹实验的。它的操作虽然并不复杂,但成本却很高,特别如果使用的是从公司购买的抗体的话。 实验讲义在文字上,主要参考了魏群主编的《分子生物学实验指导》,而在实质内容上,DNA操作部分更多的是参考了萨姆布鲁克等人的《基因克隆》第二版的中译本,蛋白质部分主要是根据自己平日工作的积累。 希望同学们能够珍惜这次机会,认真地做实验,真正地掌握些实实在在的实验操作技能,为今后的学习和工作打下基础。千万不要忽视实验技能的培养和提高,生物学的成就毕竟主要是通过实验干出来的。非变性条件下GFP的制备聚丙烯酰胺凝胶电泳
目前需要一批凝胶电泳的仪器设备,有资源的联系18366183623
有做PFGE脉冲场凝胶电泳实验的没,提供些材料。尤其是和领导建设性建议方面的。
脉冲场凝胶电泳1)高分子量DNA琼脂糖凝胶块制备和加工1.收集10ml全血,加入30ml细胞裂解液。置冰浴中至少20min直至红细胞完全溶解。2.2000r/min离心10min,移去红色上清液,再次用细胞裂解液洗涤细胞,然后用PBS重悬细胞。3.稀释单细胞悬液并取一小份用Neubauer腔计数细胞。4.用PBS重悬细胞,以达到40μl PBS中含1百万个细胞比例(1百万个二倍体哺乳动物大约含有基因组DNA 10μg)5.用PBS配制2%浓度低熔点琼脂糖溶液并保持在50℃。6.将等体积(各1ml)细胞悬液与琼脂糖溶液于室温下混匀,立即倒入凝胶块模具中。7.静置20min让琼脂糖固化,用无菌塑料杯(通常用作划菌)将凝胶块自模具中取出并置入蛋白酶缓冲液中,加入2mg/ml的蛋白酶K。8.将带有凝胶块的蛋白酶K缓冲液于50℃保持2~3天。每个盛有50ml蛋白酶缓冲液的Falcon管可容纳多达100个凝胶块。9.蛋白酶K消化后,可将凝胶块保留在此缓冲液或0.5mol/L EDTA溶液中保存于4℃。10. 此外,继续将凝胶块用高压消毒过的TE缓冲液冲洗数遍的步骤。11. 将凝胶块放入装有TE及0.04mg/ml PMSF溶液的Falcon试管中,灭活残留的蛋白酶K。12. 室温下用TE溶液漂洗凝胶块数次,将凝胶块放入另一干净的试管,可直接用于酶切反应或用0.5mol/L EDTA,(pH8.0)4℃保存凝胶块。13. 若用EDTA保存凝胶块,取出后应用TE溶液室温下漂洗30 min×2次。2)大小标准物的制备 l λ多联体1.以TE缓冲液悬浮λ多联体(Boehringer MA宝灵曼公司产品),浓度为4μg/40μl。2.用等体积TE配制的2%低熔点琼脂糖(温度保持在45℃)混匀。3.移去混合液注入预冷的凝胶块模具中。4.室温下用TE及100 mmol/L NaCl溶液温育2天。l 酵母染色体1.从YPD(酵母提取物,蛋白胨和葡萄糖)培养基瓶皿中挑选单一克隆加入10ml YPD预培养的肉汤中,30℃下剧烈震荡生长24小时,然后加入200ml YPD肉汤,剧烈振荡24~48h(产量大约100块)。2.4000×g转速,离心10min,然后用50mmol/L EDTA/10 mmol/L Tris-HCl(pH7.5)溶液悬浮。3.仍以4000×g转速离心10min,再用SCE[1 mol/L 山梨醇,0.1mol/L枸椽酸钠,pH5.8及10 mmol/L EDTA (pH7.5)]溶液重悬。4.取稀释后的细胞悬液用Neubauer腔计数。5.溶液短暂离心后,再用SCE重悬细胞,使40μl SCE溶液中含5×107个细胞(相当于80μl体积的模块中含有5×107个细胞)。6.溶液与0.1mg/ml酵母扣糖酶100T和100mmol/Lβ-巯基乙醇混匀,37℃保温15~30min。7.细胞悬液与等体积的SCE配制的2%低熔点琼脂糖凝胶混匀,保持在50℃。8.将混合物移注入预冷的凝胶块模具中。9.凝胶块用含10 mmol/L二硫苏糖醇的SCE溶液37℃下振荡温育1~2小时。10. 将凝胶块移入蛋白酶缓冲液中,加入2mg/ml蛋白酶K,50℃温育48h。11. 用50 mmol/L EDTA/10 mmol/L Tris-HCl(pH7.5)溶液洗涤凝胶块3次,每次20min。12. 凝胶块可用此混合液于4℃保存或不用漂洗直接装载入凝胶中。3)琼脂糖凝胶块中DNA的限制性内切酶消化1.应使用消毒溶液及戴无菌手套以免DNA降解。2.在Falcon试管中用1×TE溶液漂洗凝胶块20min 3次,以去除EDTA。3.混合:酶反应缓冲液(高、中或低盐缓冲液),100 mmol/L亚精胺(只用于高盐缓冲液状态),10~20单位的内切酶。20单位的内切酶就足以过夜完全消化10μg DNA。4.设立一个除内切酶成分外含有混合物各组分的阴性对照,以检查是否有非特异性DNA降解。5.将琼脂糖凝胶块加入反应混合溶液中:通常用消毒过的手术刀或套环将凝胶块移入。6.若两种内切酶所需缓冲液条件一致,可以同时或先后用两种不同的酶进行消化(先用低盐缓冲液的酶消化,再调整盐浓度)。倘若首次消化的酶要求50℃条件,在第二个酶消化时要换缓冲液。7.若要进行部分消化,则首先在同一温度和反应时间用1:10稀释的酶进行尝试。4)凝胶电泳1.将0.8%的琼脂糖在0.25×TBE中熔化后冷却至50~60℃,立即注入凝胶框架中,并插入梳子。2.凝胶固化后小心地拔出齿梳,用2把无菌手术刀将DNA凝胶块上样。若用不同内切酶消化凝胶块,则取不同样品时应将手术刀片烧灼后冷却。将DNA大小标志物上样至凝胶的两旁。3.用1%液态低熔点琼脂糖凝胶(0.25×TBE配制)密封狭槽。4.若有气泡存在,用注射器驱赶气泡。5.一旦密封的低熔点琼脂糖已固化(大约10min),可将凝胶搁入腔室,并用电泳缓冲液覆盖过胶面。6.应设定合适的电压及转换时间(参考《分子医学技术》84页表8.1)并开始电泳。两个不同电泳方向的电流应相等。7.电泳结束后,凝胶用0.25×TBE配制成的EB(0.4μg/ml)染色。8.用泵自槽中排除缓冲液,续以双蒸水冲洗电泳槽。9.凝胶成像:DNA在曝光的过程中可能会形成缺口。10. 用0.25mol/L HCl漂洗凝胶30min,让DNA脱嘌呤,并有利于转移。11. 凝胶用碱(变性溶液中)变性20min,两次,续以中性溶液1~5min。12. 采用标准Southern印迹方案将DNA转印至尼龙膜上。一般来说,印迹PFGE凝胶的时间较普通凝胶印迹时间长(约48h)。
有没有做过毛细管凝胶电泳的啊?怎么制备凝胶柱啊?第一次做,而且没人指导,文献里也没找到详细的方法,各位帮帮忙啊。急啊!
有人做过毛细管凝胶电泳吗?凝胶毛细管柱是买的还是自己做的?能提供比较好的凝胶毛细管柱制备方法吗?
美国Alpha Innotech 的凝胶电泳成像系统的camera卡的驱动,使用的是Chemilmager TM IS 4400软件,仪器上写的是 Multilmage Light cabinet ,需要的驱动是仪器上那个卡的驱动,叫着 ITI Image capture device,请使用相同仪器的兄弟姐妹们给我发过来一个吧,愁死了,仪器没法用了
关键词:单细胞凝胶电泳目的:为便于各室单细胞凝胶电泳试验结果的可比性背景知识:略原理:在细胞核中,DNA是环状附着在核基质上,细胞裂解过程中,核基质被溶解、抽提,DNA的结构则未发生变化。如果DNA链上存在缺口,则使DNA超螺旋变的松弛,DNA环向外展,同时由于暴露了阴电荷,在电场力的作用下,松动的DNA环向阳极迁移,但是由于这种松动的DNA环一端仍附着于核DNA,其迁移距离受到限制,因此尾长并不总是真实反映链缺口的多少。实际应当依靠尾长与尾部的荧光强度同时来进行分析。主体内容:操作步骤见下文主要参考文献:略操作步骤:1. 分离制备单细胞悬液:(1) 体外培养的细胞株:用胰酶消化,吹打成单细胞悬液(2) 体内脏器细胞:处死动物,取出脏器,于Hanks’液中制备成单个细胞悬液。2. 胶板制备:(1) 取20~50μl于56℃水浴中保温的0.5%普通熔点琼脂糖,铺于磨沙载玻片上,形成底胶。(2) 取100~150μl 0.5%普通熔点琼脂糖加在底胶上,再于其上加盖玻片,4℃冷凝10分钟。(3) 取下盖片,取50~100μl于37℃水浴中保温的1.0%的低熔点琼脂糖与50~100μl细胞悬液(105个细胞/ml)混匀,立即铺片,加上盖玻片,4℃冷凝10分钟。(4) 去掉盖玻片,取70~100μl于37℃水浴中保温的0.5%的低熔点琼脂糖铺片,加盖玻片,4℃冷凝。3. 细胞裂解与电泳:(1) 将制备好的胶板去掉盖玻片后,浸于4℃预冷的细胞裂解液中,4℃裂解1小时。(2) 取出胶板,放入电泳槽中,浸泡在电泳液中解旋20分钟。(3) 4℃电泳20分钟(25V,300mA)。4. 中和与染色:(1) 电泳结束,将胶板浸泡于中和液中,每次15分钟,共中和两次,注意更换中和液。(2) 取出胶板,置于染色架上,滴加5μg/ml的PI,暗处染色20分钟。(3) 蒸馏水脱色15分钟。5. 镜检和分析:(1) 在荧光显微镜下观察,绿光激发吸收滤片590nm。必要时照相记录。(2) 记数观察的细胞,记录彗星细胞出现的频率,用目镜测微尺测头长与全长,计算核DNA迁移距离。* * * * *使用两层凝胶法,经裂解、DNA解旋、电泳和中和得到湿琼脂糖凝胶片。将湿琼脂糖凝胶片置于冰冷无水乙醇中脱水10分钟,后置于空气中自发干燥。每人制备2张琼脂糖凝胶片。全部操作在采血后8小时内完成,操作过程中注意避光。脱水干燥的琼脂糖凝胶片装于含有干燥剂的载片盒中运回实验室。使用50μl 30μM的溴乙锭溶液染色、照相。使用单细胞凝胶电泳软件分析所有照片,每人随机测量100个以上细胞的尾长和olive尾矩,以尾长和olive尾矩的算术均数代表个体DNA损伤情况。
毛细管凝胶电泳缓冲液加入有机试剂如甲醇的作用原理是什么?
欢迎gl19860312担任生命科学仪器-凝胶电泳及成像版主!我们希望有更多的热心用户能加入到版主队伍中来,也希望在职的版主能在版面中发现有能力的热心用户推荐给我们。论坛正在招募版主,有兴趣的用户请参见这个帖子:http://www.instrument.com.cn/bbs/shtml/20071101/1042199/
美国Alpha Innotech 的凝胶电泳成像系统的camera卡的驱动,使用的是Chemilmager TM IS 4400软件,仪器上写的是 Multilmage Light cabinet ,需要的驱动是仪器上那个卡的驱动,叫着 ITI Image capture device,请使用相同仪器的兄弟姐妹们给我发过来一个吧,愁死了,仪器没法用了
蛋白质纯化 蛋白质分离纯化是用生物工程下游技术从混合物之当中分离纯化出所需要得目的蛋白质的方法。 是当代生物产业当中的核心技术。该技术难度、成本均高;例如一个生物药品的成本75%都花在下游蛋白质分离纯化当中。常用技术有: 1、沉淀, 2、电泳:蛋白质在高于或低于其等电点的溶液中是带电的,在电场中能向电场的正极或负极移动。根据支撑物不同,有薄膜电泳、凝胶电泳等。 3、透析:利用透析袋把大分子蛋白质与小分子化合物分开的方法。 4、层析: a.离子交换层析,利用蛋白质的两性游离性质,在某一特定PH时,各蛋白质的电荷量及性质不同,故可以通过离子交换层析得以分离。如阴离子交换层析,含负电量小的蛋白质首先被洗脱下来。 b.分子筛,又称凝胶过滤。小分子蛋白质进入孔内,滞留时间长,大分子蛋白质不能时入孔内而径直流出。 5、超速离心:既可以用来分离纯化蛋白质也可以用作测定蛋白质的分子量。不同蛋白质其密度与形态各不相同而分开。
网上都是SDS-PAGE的配方表,可是我要做非变性的电泳,请问哪位大侠有 非变性 凝胶电泳的 分离胶 和 浓缩胶 配方表啊???请赐予 万分感谢
脉冲场凝胶电泳以其重复性好、分辨力强而被誉为细菌分子分型技术的“金标准”。它可以用于的分子DNA的分离,其分辨范围可达10Mb。PFGE选用识别稀有酶切位点的限制性内切酶切割基因组DNA,获得的DNA大片段在外加脉冲电场的低浓度琼脂糖凝胶中分离,产生数量有限的DNA条带。其原理是DNA分子在脉冲电场中随着电泳方向的改变不断改变其分子构象,挤过凝胶间隙。小的DNA分子比大的分子重新定向快,在凝胶中移动快,从而使小分子DNA片段与大片段相分离,同时较大的DNA片段也能被有效分离,在凝胶上按染色体片段长度的不同而呈现出电泳带型。







 我要推广仪器
我要推广仪器
 下载APP
下载APP