推荐厂家
暂无
暂无
 留言咨询
留言咨询
 400-860-5168转1218
留言咨询
留言咨询
400-860-5168转1218
留言咨询
留言咨询
 400-860-5168转4819
留言咨询
400-860-5168转4819
留言咨询
 400-807-5250
留言咨询
400-807-5250
留言咨询
 400-860-8560
留言咨询
400-860-8560
留言咨询




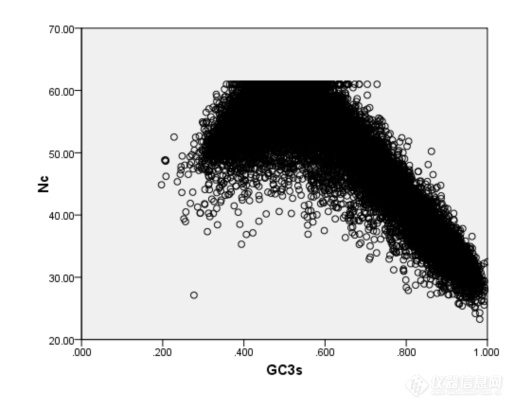
[align=center]短柄草全基因组密码子用法分析分析[/align]摘要:本研究运用CodonW程序分析了短柄草全基因组的密码子使用特性,并且通过对应分析探讨了若干重要因子对短柄草全基因组序列密码子用法的影响。结果表明短柄草基因组存在高[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]含量和低[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]含量的基因,它们在密码子使用上差异较大。Nc-plot曲线表明基因组的密码子组成受到碱基组成的影响;对应分析显示,在DNA水平上发生的核苷酸突变可能是造成短柄草基因组密码子使用偏好的主要因素;同时,基因长度和蛋白质疏水性对密码子的使用也存在一定偏性,但影响程度不大。确定了UUC等27个以G或C碱基结尾的密码子为“最优密码子”,研究结果可为短柄草基因的鉴定、表达、结构、功能等的深入研究提供参考。关键词:同义密码子偏好性,短柄草基因组,对应分析近年来,随着分子生物学的快速发展,许多小基因组的低等生物和高等模式生物的全基因组序列均被测定,为利用生物信息学方法挖掘海量基因组数据提供了便利。密码子是生物体内遗传信息传递的基本环节,是核酸携带信息和蛋白质携带信息间对应的基本规则。在长期进化过程中,任一物种的基因都会逐渐适应宿主的基因组环境,而形成特定的且符合宿主基因组的密码子用法,因此不同生物具有不同的密码子使用模式。以生物基因组数据为基础,研究其密码子使用模式,为深入研究基因的结构、功能和基因组进化,以及指导基因转化等具有重要意义。密码子具有简并性,生物在同义密码子的使用上并不是完全随机的,而是具有一定的偏向性,对有的密码子使用频率高,有的使用频率低,甚至避免使用,这种不均衡使用密码子的现象普遍存在于原核和真核生物中。早在20世纪70年代,人们在研究基因的异源表达时,就已经意识到密码子偏性的重要性[1],随着不同生物基因组数据的获得和各种数据库的构建,更多的研究者对密码子偏性的研究产生了浓厚的兴趣,尤其在分子进化,翻译调控等研究领域,通过对不同物种的密码子使用偏性的大量研究[2~4],发现不同物种的基因在密码子使用上存在着明显的偏性。 短柄草是一种广泛分布于温带地区的禾本科植物,与小麦,大麦和燕麦同属早熟禾亚科,原产于非洲北部,欧洲南部和亚洲中部,包含约10个亚种。该植物为一年生,自花授粉,植株高度15~20cm,生育期70~80d,柄草植株较小,适应性强,不象种植水稻那样需要严格的生长条件。生育期短,籽粒产量较高,一年可以繁殖4~5代,繁殖系数达140左右。未成熟胚和成熟胚愈伤组织诱导率高,农杆菌介导和基因枪介导的转化体系已经建立,胚性愈伤组织分化率90%以上,转化效率最高可达55%左右。基因组小,染色体少,DNA重复序列低,获得突变体容易,突变性状容易显现,具备了模式植物的所有基本特征。加之短柄草基因组序列与黑草麦,小麦,大麦等早熟禾亚科植物高度相似,很多重要农艺性状与温带禾草类植物相似,如株型,穗型,粒型,抗逆性,生长习性和病原菌等,其中麦类作物白粉病菌,条锈病菌和稻类作物瘟病菌都可侵染短柄草植株,引起相应症状[7]。其籽粒不含高分子量麦谷蛋白亚基,低分子量麦谷蛋白亚基也很少,并与小麦一样具有二倍体,四倍体和六倍体,因此短柄草是小麦等基因组庞大的重要农作物理想的模式植物,借此来获得目前小麦等早熟禾类植物中尚缺少的遗传信息和基因共线区,进而对小麦等重要植物进行基因定位,克隆,突变,测序和功能等方面的研究[8]。 目前,在短柄草的生物学、细胞学和遗传学特性方面开展了大量研究,并且其全基因组测序也基本完成[9],为深入研究其密码子用法提供了便利。因此本研究将以短柄草全基因组序列为基础,分析其基因的密码子用法特性和影响密码子使用的因素等,其研究结果将对指导转基因及对基因进行特定分子改造,提高其在短柄草中的表达效率和完善基因预测软件,提高基因预测和基因组注释准确性等均具有重要的参考价值,同时也为深入开展基因结构和功能,分子进化等研究提供理论基础。1.实验材料与方法1.1材料 短柄草全基因组DNA序列来源于短柄草官方数据库(http://www.brachypodium.org/node/8),根据基因组序列的注释信息,获得蛋白编码基因序列,为了减少长度较短的基因变异带来的样本误差,根据国际惯例,去除小于300bp的基因,去除中间不表达的密码子,终止密码子。编写程序提取剩下的蛋白编码基因的CDS(coding sequence)序列。1.2方法用codonw软件计算短柄草全基因组的密码子用法相关参数,主要包括有效密码子数(Effective Number of Codon,ENC)、基因的G+C含量([url=https://insevent.instrument.com.cn/t/Mp]gc[/url]%)、[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]3s%、相对同义密码子使用度(relative synonymous codon usage,RSCU)、氨基酸组分指数(平均亲水性值(gravy))、基因长度即氨基酸数(L_aa)。其中,有效密码子数(Effective Number of Codon,ENC)描述密码子使用偏离随机选择的程度,能反映密码子家族中同义密码子的非均衡性的偏好;其取值范围在20到61之间,即如果每种氨基酸只使用一种密码子则有效密码子数为20,如果各种同义密码子的使用机会完全均等,则有效密码子数为61,数值越小偏性越强。此值是以描述密码子使用偏离随机选择的程度,能反映密码子家族中同义密码子的非均衡性的偏好。基因密码子偏爱程度越大,ENC值越小。RSCU是指对于某种特定的密码子在编码对应氨基酸的同义密码子间的相对频率;[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]3s%表示同义密码子第三位碱基的G+C的含量。为进一步了解该家族基因密码子使用特征和影响密码子使用的因素,对7个基因的相对同义密码子使用度进行了对应性分析(correspondence of analysis,COA)。2 结果与分析2.1 基因的碱基组成对密码子使用的影响图一 短柄草基因NC值散点图[img=,515,409]https://ng1.17img.cn/bbsfiles/images/2019/10/201910311236371230_3093_3295053_3.png!w515x409.jpg[/img]2.2短柄草基因密码子使用特性的对应性分析[img=,690,535]https://ng1.17img.cn/bbsfiles/images/2019/10/201910311237226440_1452_3295053_3.png!w690x535.jpg[/img][img=,690,534]https://ng1.17img.cn/bbsfiles/images/2019/10/201910311237233450_935_3295053_3.png!w690x534.jpg[/img]2.3 确定最优密码子Phe UUU 0.05 (323) 1.23 (19733) Ser UCU 0.22 (990) 1.60 (23834) UUC* 1.95 (13527) 0.77 (12294) UCC* 2.55 (11715) 0.64 (9499) Leu UUA 0.02 ( 93) 0.83 (11755) UCA 0.14 (629) 1.52 (22651) UUG 0.16 (1003) 1.37 (19558) UCG* 1.53 (7023) 0.35 (5159) CUU 0.14 (847) 1.55 (21987) Pro CCU 0.22 (1306) 1.57 (17584) CUC* 3.38 (20676) 0.61 (8661) CCC* 1.35 (7940) 0.47 (5299) CUA 0.07 (452) 0.70 (9983) CCA 0.20 (1184) 1.62 (18078) CUG* 2.23 (13637) 0.94 (13401) CCG* 2.22 (13058) 0.34 (3792) Ile AUU 0.12 (398) 1.41 (21216) Thr ACU 0.10 (401) 1.46 (16515) AUC* 2.76 (9124) 0.70 (10557) ACC* 1.75 (7291) 0.66 (7397) AUA 0.12 (380) 0.89 (13461) ACA 0.12 (509) 1.56 (17636) Met AUG 1.00 (8512) 1.00 (20892) ACG* 2.03 (8478) 0.32 (3563) Val GUU 0.10 (693) 1.67 (23852) Ala [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]U 0.14 (1914) 1.65 (26184) GUC* 1.71 (12491) 0.63 (9025) [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]C* 1.98 (27398) 0.58 (9131) GUA 0.05 (349) 0.75 (10713) [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]A 0.13 (1802) 1.48 (23459) GUG* 2.14 (15605) 0.95 (13562) [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]G* 1.75 (24170) 0.29 (4678) Tyr UAU 0.05 (229) 1.28 (14480) Cys UGU 0.06 (194) 1.10 (9360) UAC* 1.95 (8126) 0.72 (8075) U[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]* 1.94 (6645) 0.90 (7595) TER UAA 0.42 (172) 0.82 (335) TER UGA 1.63 (665) 1.30 (530) UAG 0.94 (384) 0.87 (356) Trp UGG 1.00 (4992) 1.00 (10053) His CAU 0.15 (598) 1.42 (16785) Arg CGU 0.16 (750) 0.85 (6945) CAC* 1.85 (7568) 0.58 (6825) C[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]* 2.75 (12565) 0.49 (4043) Gln CAA 0.15 (627) 1.05 (20215) CGA 0.11 (500) 0.64 (5273) CAG* 1.85 (7975) 0.95 (18259) CGG* 1.92 (8761) 0.55 (4527) Asn AAU 0.12 (465) 1.31 (26650) Ser AGU 0.05 (235) 1.13 (16754) AAC* 1.88 (7141) 0.69 (13985) A[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]* 1.52 (7002) 0.77 (11441) Lys AAA 0.11 (552) 0.98 (27077) Arg AGA 0.10 (445) 1.94 (15854) AAG* 1.89 (9406) 1.02 (28423) AGG 0.96 (4387) 1.53 (12516) Asp GAU 0.15 (1344) 1.44 (39136) Gly GGU 0.11 (882) 1.34 (18423) GAC* 1.85 (16539) 0.56 (15322) G[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]* 2.53 (20795) 0.71 (9826) Glu GAA 0.17 (1437) 1.13 (36292) GGA 0.19 (1522) 1.26 (17423) GAG* 1.83 (15812) 0.87 (27746) GGG* 1.18 (9700) 0.69 (9476) 注:Number of codons in high bias dataset 372333 Number of codons in low bias dataset 915109标注*的密码子是(p 0.01)3 讨论密码子使用偏好是突变偏好、自然选择和遗传漂变等共同作用的结果,与碱基组成、翻译选择压力、基因表达水平、基因长度、蛋白质氨基酸组成、碱基突变频率和模式、mRNA二级结构稳定性等很多因素有关[17]。张晓峰[18]等研究表明,单子叶植物基因组的[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]含量在同义密码子使用偏性的产生过程中起着决定性的作用,同义密码子使用偏性强烈的基因往往偏爱使用C或G结尾的密码子,且第三位密码子突变往往是密码子偏好性发生变化的决定原因。短柄草基因密码子使用模式的调查表明其中有高含量的[url=https://insevent.instrument.com.cn/t/Mp]gc[/url],并且[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]3的含量高于[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]1和[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]2。这表明相对于以A和T结尾的密码子而言,这些基因偏好于使用以G或C结尾的密码子。从原核生物到真核生物的基因中,密码子使用偏好是一个被广泛研究的重要进化现象。研究发现,许多因素,比如碱基组成,基因表达水平,蛋白质疏水性等影响着密码子的使用。为了解释密码子使用偏好的起因,也有许多假设被提了出来。其中被广为接受理论是“选择——突变——漂移”模型。该模型认为在对偏好密码子的选择和通过突变-漂移对非偏好密码子的保留之间,同义密码子的使用偏性存在一种平衡。本文的研究结果显示,[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]3s值与ENC值密切相关,并且基因也位于第一轴线,揭示了碱基组成是影响短柄草基因组中的密码子使用偏好的主要因素。碱基组成是影响短柄草基因密码子使用的主要因素,基因长度和蛋白质的疏水性在短柄草基因密码子使用中也起到了一定的作用,相似的结果在水稻、小麦中被发现[15,19]。本研究发现,在基因长度和[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]之间存在很强的负相关性。这表明,高[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]含量的基因越短,密码子偏好就越大。可能的原因是富含AT基因的翻译效率比富含[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]基因的翻译效率更高,这种效率的差异对长的基因更为重要。通常,全基因组的基因表达值在许多多细胞真核生物中并不能得到,特别是基因表达水平在不同的组织和不同发育阶段不一样时。因此,要定量相当困难。在短柄草基因组中,目前还缺少相当数量的基因表达的准确数据。另外,我们发现[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]含量特别是在第三个碱基位置的[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]含量较大的影响着密码子的偏好时,暗示着碱基突变可能是重要因素,同时,碱基突变又受控于翻译选择。所以,尽管基因表达水平影响着密码子的使用,但这影响还是远远小于核苷酸组成对密码子使用的影响。因此,我们没有进一步分析基因表达的影响。通过优化密码子,提高外源基因在微生物、植物、动物中的表达已有不少成功报道,而确定最优密码子可为合理有效进行密码子改造提供可靠信息。本文确定了UUC等27个密码子为短柄草全基因组的最优密码子。分析结果可为指导转基因及对基因进行特定分子改造,提高其在短柄草中的表达效率和完善基因预测软件,提高基因预测和基因组注释准确性等提供重要的参考价值。参考文献[1] Stanley D,Farnden K J F, MacRae E A. Plant a-amylases:Func-tions and roles in carbohydrate metabolism[J]. Biologia,Bratislava,2005.60(suppl l6):65-71[2] Smith AM. Zeeman SC, Smith S M. Starch degradation[J]. Annu Rev Plant Biol,2005,56(25):73-98[3] Asatsuma S, Sawada C, Itoh K et al. Involvement of α-amylase I-1 in starch degradation in rice chloroplasts[J]. Plant Cell Physiol,2005,4:858-869[4] Kaplan F, Guy C L. β-amylase induction and the protective role of maltose during temperature shock[J]. Plant Physiol, 2004, 1:1674-1684 [5] Kaplan F,Guy C L. RNA interference of Arabidopsis beta-amylase 8 prevents maitose accumulation upon cold shock and increases sensitivity of PSII photochem-ical efficiency to freezing stress[J]. Plant J.2005,44(13):730-743[6] Joho Mundy, Anders Brandt. Messenger RNAs from the Scutellum and Aleurone of Germinating Barley Encode (lm3,14)--D-Glucanase, a-Amylase and Carboxypeptidase[J]. Plant Physiol, 1985,79(5):867-871 [7] 言普,李桂双.高压对水稻种子细胞膜透性和淀粉酶活性的影响[J]. 浙江大学学报(农业与生命科学版),2007,33(5):174-179[8] Monica M, Sanwo and Darleen A. DeMason. Characteristics of a-Amylase during Germination of Two High-Sugar Sweet Corn Cultivars of Zea mays L[J]. Plant Physiol, 1992,99(8):1184-1192[9] Goldman N , Yang Z. A codon based model of nucleotide substitution for protein coding DNA sequences[J]. Molecular Biology and Evolution,1994,11(9):725-736[10] Schmidt W. Phylogeny reconstruction for protein sequences based on amino acid properties[J]. Mol Evol,1995,41(8) :522-530[11] 时成波, 吕安国.改造稀有密码子提高SEA蛋白表达量[J]. 生物工程学报,2002,18(4):477-480[12] Ghosh T C , Gupta S K, Majumdar S. Studies on codon usage in Entamoeba histolytica[J]. Int J Parasitol,2000,30(6): 715-722[13] Musto H, Cruveiller S. Translational selection on codon usage in Xenopus laevis[J].Molecular Biology and Evolution,2001,18(9):1703-1707[14] 廖登群,张洪亮等. 水稻(Oryza sativa L.)a-淀粉酶基因的进化及组织表达模式[J]. 中国农业大学学报,2009,14(5):1-11[15]刘汉梅,何瑞. 玉米密码子用法分析[J]. 核农学报,2008,22(2):141-147[16] Jia M, Luo L. The relation between Mrna folding and protein structure[J]. Biophys Res Commum, 2006,343(4):177-182[17] 赵耀,刘汉梅. 玉米waxy基因密码子偏好性分析[J]. 玉米科学,2008,16(2):16-21 [18] Wang H C,Hickey D A. Rapid divergence of codon usage patterns within the rice genome[J].BMC Evol Biol,2007,15(8):347-356
http://img.dxycdn.com/trademd/upload/userfiles/image/2013/01/B1357710940_small.jpg梅花因其独特的花香,在很多诗词中成为人们吟诵的对象。那么,它的花香到底来自何处呢?我国科学家从基因组水平,揭示了合成梅花花香中重要成分乙酸苯甲酯的BEAT基因家族34个成员,并构建完成了首张梅花全基因组精细图谱。其研究论文在2012年12月27日《自然—通讯》亮点论文在线发表。我国梅花基因组项目首席专家、北京林业大学教授张启翔率领项目组,选取位于梅花起源中心的西藏野生梅花进行基因组测序,从基因组水平,揭示了合成梅花花香中重要成分乙酸苯甲酯的BEAT基因家族34个成员,在梅花基因组中显著扩增并且其中12个成员串联重复分布,从而使梅花具有独特的花香;推测梅花基因组中6个串联重复的DAM基因和其上游过多的CBF结合位点是梅花提早解除休眠的关键因子,从而解释“踏雪寻梅”之说。张启翔告诉记者,梅花全基因组测序的完成以及高密度遗传图谱构建,有助于揭示梅花花期早、花香独特等重要观赏性状的遗传基础,有助于挖掘与诸多重要性状相关的功能基因,为今后进一步揭示梅花花期、抗病调控机制、梅花及相关种属的分子育种奠定基础。研究中,项目组还揭示了蔷薇科植物进化规律。张启翔说,通过分析梅花的进化发现,梅与苹果发生分化后,并没有出现近期的全基因组复制事件,同时结合已完成的苹果和草莓基因组序列,成功重建了蔷薇科9条原始染色体,揭示了蔷薇科植物进化规律,为开展蔷薇科物种比较基因组学研究奠定重要的理论基础。据介绍,该科研成果由北京林业大学、深圳华大基因研究院及北京林福科源花卉有限公司等多家单位合作完成。目前,转录组数据组装及基因功能注释数据已在相关网站对外公开。
由来自中国、美国、荷兰、以色列等14个国家的300多位科学家组成的“番茄基因组研究国际协作组”,历时8年多的艰苦努力,于近日完成了对栽培番茄全基因组的精细序列分析。今天,国际权威学术期刊《自然》以封面文章发表了这项重大科学成果。 番茄是研究果实发育的经典模式植物,我国科学家在这项国际番茄基因组研究中作出了重要贡献。作为中方协调人,中科院遗传与发育生物学研究所研究员李传友和薛勇彪负责第3号染色体的测序工作,中国农科院蔬菜花卉研究所研究员黄三文和杜永臣负责第11号染色体的测序工作。番茄基因组有12条染色体,中国科学家高质量地完成了番茄基因组测序总任务的1/6,标志着我国成为番茄基因组学研究的强国之一。 8年来,国际协作组采用“克隆连克隆”和“全基因组鸟枪法”相结合的测序策略,在解码的番茄基因组中,共鉴定出约34727个基因,其中97.4% (33840个)的基因已经精确定位到染色体上。番茄基因组的解读,是科学家通过国际合作完成的又一个高质量的模式植物的基因组序列分析,对于不同物种之间的比较基因组学研究具有重要价值,这项工作将极大推动番茄乃至包括马铃薯、辣椒、茄子等在内的茄科植物的功能基因组研究,为培育具有高产、优质、抗病虫害、抗逆等优良性状的番茄新品种打下了良好的基础,对推动全世界的番茄生产具有重要意义。 有关专家表示,我国蔬菜种业面临着强大的国际竞争。中国在国际蔬菜基因组研究领域具有优势地位,而如何把基础科研的优势转化为产业优势,是目前面临的主要挑战。科学家建议,应在进一步巩固蔬菜基因组研究优势的基础上,加强蔬菜作物分子设计育种体系的建设,并与常规育种相结合,加速有自主知识产权优良品种的培育,这对于支撑我国蔬菜产业可持续发展、提升我国蔬菜种业的国际竞争力具有重要意义,也是不可错过的历史机遇。








 我要推广仪器
我要推广仪器
 下载APP
下载APP