推荐厂家
暂无
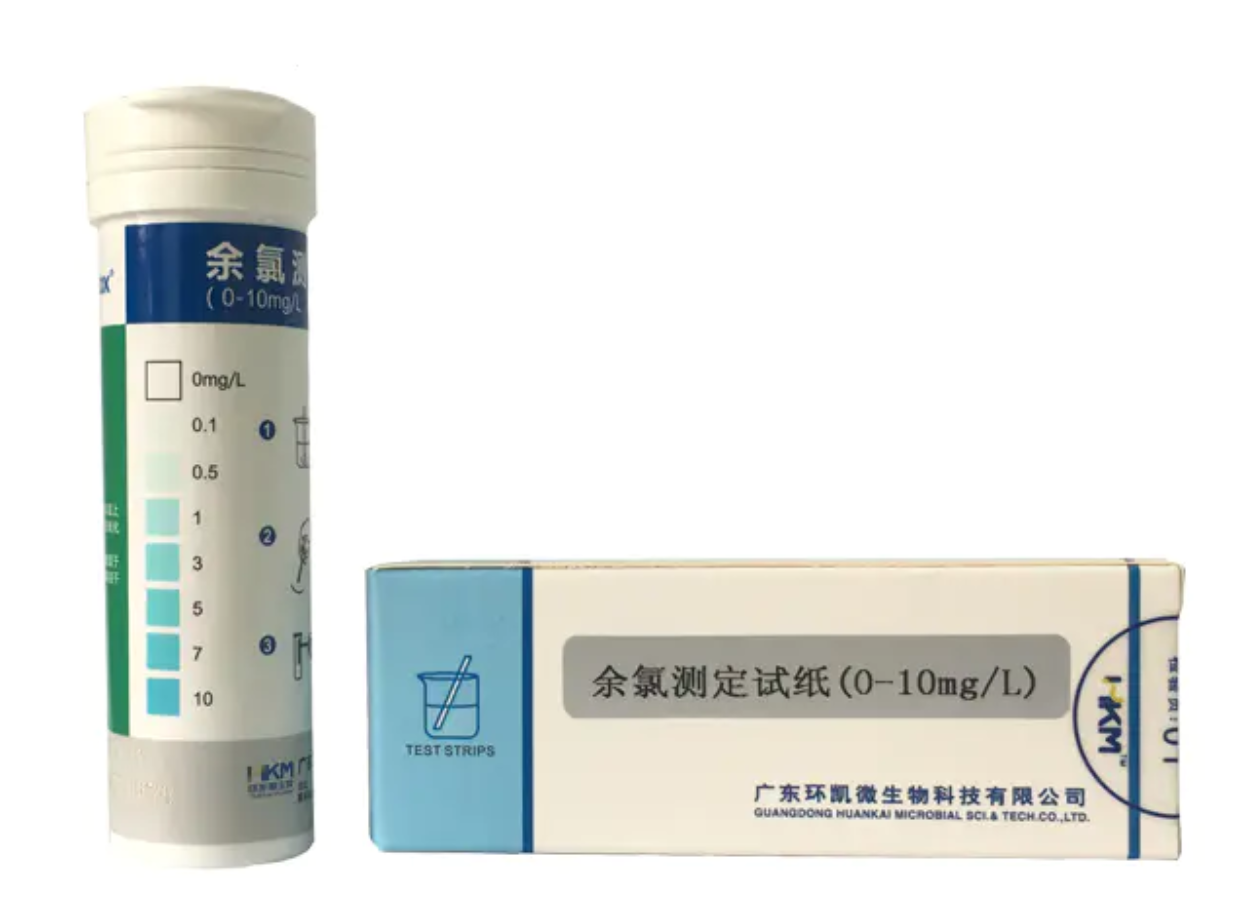
暂无




[b]关于公开征求《关于促进土壤污染绿色低碳风险管控和修复的指导意见(征求意见稿)》意见的通知[/b] 为贯彻落实党的二十大精神,加快土壤污染风险管控和修复领域绿色化低碳化发展,按照《中华人民共和国土壤污染防治法》《“十四五”生态环境保护规划》《减污降碳协同增效实施方案》要求,我部组织起草了《关于促进土壤污染绿色低碳风险管控和修复的指导意见(征求意见稿)》,现公开征求意见。征求意见稿及其起草说明可登录我部网站(http://www.mee.gov.cn/)“意见征集”栏目检索查阅。 各机关团体、行业协会、企事业单位和个人均可提出意见和建议。有关意见和建议请书面反馈我部,电子文档请同时发至联系人邮箱。征求意见截止时间为2023年7月10日。 联系人:生态环境部土壤生态环境司 任静、常方方 电话:(010)65645698、65645693 传真:(010)65645732 邮箱:wrdk@mee.gov.cn 地址:北京市东城区东长安街12号 邮政编码:100006 附件:1.[url=https://www.mee.gov.cn/xxgk2018/xxgk/xxgk06/202306/W020230626547396105697.pdf]征求意见单位名单[/url] 2.[url=https://www.mee.gov.cn/xxgk2018/xxgk/xxgk06/202306/W020230626547396309700.pdf]关于促进土壤污染绿色低碳风险管控和修复的指导意见(征求意见稿)[/url] 3.[url=https://www.mee.gov.cn/xxgk2018/xxgk/xxgk06/202306/W020230626547396885764.pdf]《关于促进土壤污染绿色低碳风险管控和修复的指导意见(征求意见稿)》起草说明[/url][align=right] 生态环境部办公厅[/align][align=right] 2023年6月24日[/align] (此件社会公开)
工艺变更指导意见2008(工艺核查要点〈08年工艺核查指导〉)本指导意见适用于制剂、原料药生产工艺变更。一、概述工艺变更必须符合《药品注册管理办法》的有关要求。任何工艺变更,我们都必须评估变更对产品的影响,并作出评估报告。本指导意见提供了工艺变更研究工作的基本内容和一般性技术要求,阐述了研究工作需要重点关注的问题和研究工作的基本思路。企业可参照本指导意见的基本思路,参考国内外有关技术指导原则,对各种具体变更进行全面深入的研究。二、基本原则企业应在变更前后样品质量、稳定性、生物学等方面进行全面研究验证的基础上,还需注意对研究结果进行全面的分析和评估,论证变更对产品品质的影响,即变更前后产品质量是否等同,临床治疗是否等效。研究工作一般应考虑进行以下方面:1、评估变更对药品的影响产品发生变更后,需通过一定的研究工作考察和评估变更对产品质量、安全性、有效性的影响,包括对产品化学、物理学、微生物学、生物学、生物利用度及稳定性方面任何改变进行的评估。研究工作宜根据变更的具体情况和变更的类别、原料药或制剂的性质,及变更对产品影响程度等综合考虑确定。例如,对于变更前后产品杂质变化的考察,宜首先选择或建立合理的色谱方法,对变更前后杂质情况(杂质个数、杂质量)进行比较性分析。如果变更后检出新的杂质或降解产物,或原有杂质水平超出标准中限度的规定,则需要考虑进行相应的毒理学研究工作。除本指导意见中各类变更项下建议进行的研究工作外,企业还需结合变更的特点及具体变更情况,选择其他重要项目进行研究。如片剂某些生产工艺变更,除溶出/释放行为比较外,还需要考察某些重要的物理参数如脆碎度等是否发生改变。2、评估变更前后产品的等同或等效性在对上述产品化学、物理学、微生物学、生物学、生物利用度及稳定性方面进行研究验证工作的基础上,进行全面的分析,评估变更对药品质量、安全性及有效性方面的影响。一般可通过对变更前后考察结果进行比较和分析,来判定变更前后结果是否等同。这些比较性研究既包括溶出度、释放度等项目的比较,也包括对药品稳定性等某一方面性质的全面比较分析。对于溶出度、释放度、生物利用度等具体项目,可选择适宜的统计学方法进行比较和分析,严格意义上,变更前后产品并不是保持完全一致,而需保持等同、等效,即产品质量等同,临床治疗等效。某些情况下,产品变更前后并不能保持等同或等效,即变更对产品质量、安全性和有效性产生一定影响。如果企业希望实施这种变更,则需要通过药学、生物学等系列研究工作,证明实施这种变更不会对产品品质产生负面影响。例如,研究发现某生产工艺变更引发新的降解产物,但进一步研究结果证实,该降解产物并不会引发安全性方面的担忧等。3、关于研究用样品的考虑企业进行工艺水平变更研究验证的样品一般应为生产规模样品,或至少在GMP车间生产的样品。变更前后产品质量比较研究(如溶出度、释放度比较实验)一般采用变更前三批生产规模样品和变更后至少一批生产规模样品或GMP车间生产的样品进行。变更后产品稳定性实验一般采用1-3批生产规模样品或GMP车间生产的样品进行3-6个月加速实验和长期留样考察,并与变更前三批生产规模样品稳定性情况进行比较。稳定性实验产品具体批次和考察时间需根据变更对产品品质的影响程度、产品的稳定性情况等因素综合确定,对于较大变更,或结果提示产品稳定性差的,应选择较多的样品批次并延长考察时间。[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=121869]工艺变更指导意见2008(工艺核查要点〈08年工艺核查指导〉)[/url]
各省、自治区、直辖市食品药品监督管理局(药品监督管理局),总后勤部卫生部药品监督管理局: 为加强药物I期临床试验的管理,提高临床试验生物样本分析实验室的分析质量,有效地保障受试者的权益与安全,确保所产生的数据和结果的可靠性、完整性和科学性,根据《药物临床试验质量管理规范》,我司组织起草了《药物Ⅰ期临床试验管理指导原则(征求意见稿)》、《药物临床试验生物样本分析实验室管理规定(征求意见稿)》。现公开征求意见,请于2011年4月30日前将修改意见反馈我司。(征求意见稿可在国家食品药品监督管理局网站下载) 联系人:蓝恭涛,唐慧鑫 电 话:010—88330742,0722 传 真:010—88363228 地 址:北京市西城区宣武门西大街26号院2号楼 邮 编:100053 E-mail:yjjdc@sda.gov.cn 附件:1.《药物Ⅰ期临床试验管理指导原则(征求意见稿)》 2.《药物临床试验生物样本分析实验室管理规定(征求意见稿)》 3.起草说明 国家食品药品监督管理局药品注册司 二○一一年三月十八日

 400-659-9826
400-659-9826
 留言咨询
留言咨询
 留言咨询
留言咨询
 留言咨询
留言咨询










 我要推广仪器
我要推广仪器
 下载APP
下载APP


