标准曲线不含零点,是指在标液浓度范围内没有0.00这个浓度/含量,自然也就没有零点这个点,而不是指标准曲线过不过原点。 从最开始接触原吸做铅镉标曲,标液浓度总是从0.00起,于是想当然地认为所有的标曲都是应该过原点。有时标曲不过原点,认为是标曲做得不好之故。直到最近发现有一些标曲中本身就不含有零点那个点,这样的标曲在HJ491-2009,GB/T 7475-1987,GB/T 5413.21-1997中都有提到。 为什么有的标曲包含零点,有的却否?我从最近做了一条浓度大跨度的铬标准曲线上得到了答案。浓度范围大了以后,线上的所有点组成的不再是直线,而是一条二次线或更高级的曲线。所谓线性,只是在一个较小的区段内才可以认为呈一条直线。在较大的区段则不成直线。我们原吸通常都是用线性方程校准,因此对某些浓度较大的区段做标曲时就不能再带零点了。 从这里也可以看出,要求被测样品的浓度必须在标曲中间位置的必要性。如果样品值远离标曲浓度范围,则已不服从线性关系了,自然就不能再用那个方程计算了。 这样的结论对不对呢?似乎暗合了坛子里老师们一再要求样品值要在标曲中间的说法。发现一些别人早已知道的事实就大惊小怪,真是要贻笑大方了。反之,如果在论证过程中有错误的地方,希望得到老师们一定要给与积极的指正。
标准曲线不通过零点对结果的影响关于标准曲线不通过零点对结果的影响的原因,关于标准曲线不通过零点对结果的影响的相关知识。 王庆敏 兰州医学院第一附属医院检验科 数理医药学杂志 1998 4 全文下载不了,欢迎大家讨论!
石墨炉做“铅”时,标准曲线做了4个点,0.0ug/l、2.0ug/l、5.0ug/l、10.0ug/l、吸光度依次是0.0074、0.0099、0.0145、0.0207,回归方程y=0.00134x+0.00744,其中零点(0.0ug/l)就是配制的1%(v/v)硝酸,其它3个浓度皆是用1%(v/v)硝酸定容的,如果要把零点吸光度先扣除掉的话,吸光度就依次为0.0000、0.0025、0.0071、0.0133,那么回归方程y=0.00134x+0.00002,问题是我测的水样用第一个回归方程(含零点),做出的值都是负值,水样未经任何添加处理,就是“裸水样”,表明被测水样中的“铅”含量比零点【1%(v/v)硝酸】中的“铅”含量低,试问遇到这种情况我是不是应该用第二个回归方程y=0.00134x+0.00002,这样水样就不会显示负值,而是实际所测值,这样亡羊补牢行不行?请大家赐教,不知道我这样分析是否有道理
如题,HJ803校准曲线控制要求每分析二十个样品进行一次零点分析,要求测定值与实际浓度值的相对偏差≤30%。零点分析实际浓度值应该是0啊,ICP/MS测定零点时只要测定值非零,都达不到这个控制范围,标准要求是否有毛病?
原子吸收元素的标准曲线是不是一定要过零点,不过零点是不是有问题呢
仪器:北京恒通瑞利 7w多 火焰原子吸收问题:①最近测量Pb、Cu标液2、4、6、8µg/ml,不考虑零点线性很好,但是不过零点,偏高。此种情况正常吗?原来可以过零点的。②标准曲线绘制后,测试中,火焰的大小可调否,会影响吸光度吧。已经帮老板买了近百万的仪器了,都是在仪器信息网找的供应商资料,不过还是第一次发现有这个论坛,太好了。
标准曲线的两层意义?标准曲线是否要加入零点?哪位大神可以帮帮忙?谢谢!
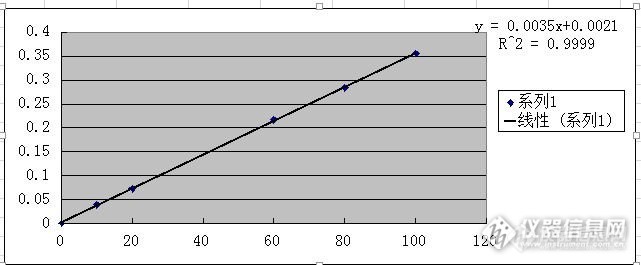
http://ng1.17img.cn/bbsfiles/images/2014/09/201409161720_514098_2205412_3.jpghttp://ng1.17img.cn/bbsfiles/images/2014/09/201409161720_514099_2205412_3.jpghttp://ng1.17img.cn/bbsfiles/images/2014/09/201409161720_514100_2205412_3.jpg做了个氨氮的标准曲线,用质控样检测,质控样的标准值为2.55+/-0.1mg/L不加零点的标准曲线为y=0.0035x+0.0035,R2=0.9999,加零点的为y=0.0035+0.0021,R2=0.9999用不加零点的回归方程算得质控样为2.56mg/L,用加零点的回归方程算得质控样为4.57mg/L,严重超出了标准值,那绘制氨氮标准曲线时,到底要不要加零点?这根曲线还能不能用啊?
你的标准曲线是否采用5个曲线点以上(不含零点)?CNAS CL10-2012里面有讲标准曲线要达到5个点以上,平时采用3个点,或者4个点的(不包括标准空白),你会如何应对?整改还是照旧呢
在做GA/T 842-2019血液中乙醇含量鉴定时,做乙醇标准曲线是否要强制过零点
用紫外分光光度计,参比溶液是纯水,标准曲线的零点是有吸光度的,我每次测试都需要重新做标准曲线吗?这样做很麻烦的。但是如果我用同一条曲线,面临的问题是,并不是每次测试零点的值都是相近的,有可能偏高或者偏低,这当如何是好?请各位高手指教。
标准曲线过不过零点有什么区别?或者说对于样品的测定有什么影响吗?
标准曲线要不要强制过零点?
请问 标准曲线靠近上边顶点的误差大还是下边零点的误差大?那标准偏差那?
《HJ 803-2016 土壤和沉积物 12种金属元素的测定 王水提取-[url=https://insevent.instrument.com.cn/t/yp][color=#3333ff]电感耦合等离子体质谱[/color][/url]法》这方法里的11.2规定了:每20个样品或每批次(少于20个样品/批)样品分析完毕后,应进行一次[b]标准曲线零点[/b]分析,其测定结果与实际浓度值的相对偏差应小于等于30%。我想知道里面的“标准曲线零点”就是曲线中零浓度的点吗?那不是硝酸吗?按理说做的越好,试剂越纯,污染越少的话浓度值不应该是零的么,而且浓度那么低,几乎没可能达到30%吧?比较来看的话水质标准里说的是“曲线最低点”,我理解其为非零点的最低点,也就是标准1,可是这土壤标准就没办法这么理解了,谁明白其中的含义,烦请赐教。
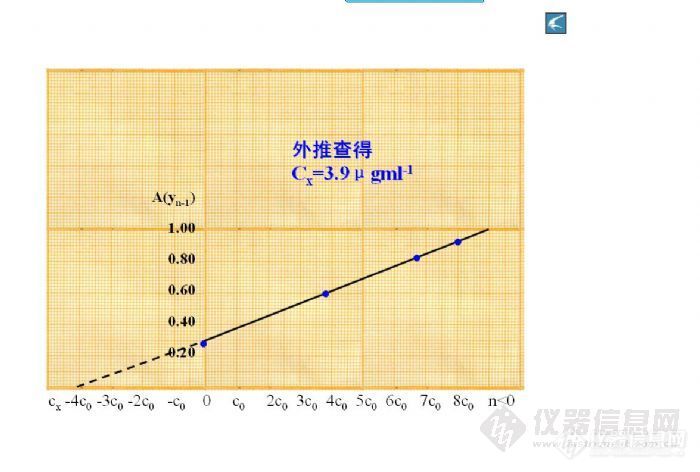
作标准曲线时,一般把不含待测元素的那个点作为零点,作校零用。而用标准加入法时,是不是不需要零点,也不需要校零?发了几个图片,是关于标准加入法的,由此得出这样的结论。看看我错在哪里?[img]http://ng1.17img.cn/bbsfiles/images/2017/01/201701191651_625950_1634383_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2017/10/2009111205225_01_1634383_3.jpg[/img]
液相检测为了验证机器的稳定性和准确性要定期做曲线,国标要求过零点,我个人理解是机器可以强制过零点,就可以不做零标品了,大家认为呢?
关于空白和零点的问题在ICP测试过程中,我们经常会遇到关于空白和零点的问题。在方法选择时,一般采用高纯物质打底+标液配制曲线或用几个国家标准物质。关于曲线零点的问题,相关手册上一般是要求含等量基体但不含待测元素(这不现实)。思路:1 零点用超纯水做。2 零点用高纯物质做。(但高纯物质一般99.95%左右,可能存在微量的待测元素)3 零点用试剂空白做。讨论:关于1,超纯水可能是含量最低的,峰型调整好,待测元素一般不存在负值。(但也不是绝对,不同元素基体所导致的基线漂移带来的强度对各个元素的影响不同。)关于2,一般找不到含等量基体但不含待测元素的高纯物质,那在用这个打底的时候,是否要用其它方法测出基体中的待测元素含量呢?(外标法或标准加入)关于3,一般在配制标液时,都要求做酸度匹配。加入的酸中如果存在少量的待测元素,我们应该如何应对呢?(是否需要提前测出加入酸中待测元素的含量?)
电阻、电容、电感是电子线路中必定使用的零部件。在进行电子线路的设计的基础上,准确地测量这些零部件的值是极其重要的。测量这些零部件的值,一般使用LCR测试仪。LCR测试仪不仅能自动判断元件性质,而且能将符号图形显示出来,并显示出其值,还能测量Q、D、Z、Lp、Ls、Cp、Cs、Kp、Ks等参数,且显示出等效电路图形。用LCR测试仪来测量零部件时,误差是难免的,一般我们有两种校正。 其一就是,零点校正。当LCR测试仪的零点漂移对于测量值不能忽略时,就需要进行零点校正。因为零点漂移会随着电缆和电极的物理配置不同而变化,所以进行开路和闭路的零点校正时,必须与连接零部件时的电缆布线、电极间隔等相同。 其二就是,负荷校正。为了进行负荷校正,首先需要准备好标准器具或者已知准确值的零部件。在进行了零点校正之后,再测量已知准确值的标准阻抗Zstd,如果得到的测量值为Zms,那么就按照以下公式来求出校正系数。LCR测试仪除了测量夹具等不同所引起的零点漂移以外,如果还有不能够忽略的测量误差,那么可以进行负荷校正,以提高测量精确度。即使对于没有负荷校正功能的LCR测试仪 ,也能够对各个阻抗量程和频率求取校正系数,自己进行校正。
水质监测中用比色分析法,要做标准曲线,零点是否需要带入回归?
ICP-AES做标准曲线时,曲线原点不通过零点,多数情况是低于零点,但线性很好,做出结果偏低。请问这个问题如何解决?
请问浊度仪零点用空气校准和用零浊度液校准的区别。哪个更好一些。
零点校准和终点校准分别是通过哪些方式进行的,零点校准和终点校准需要每次检测都要进行吗?
请高手指点一下有关帕纳科衍射仪零点校准的方法以及具体步骤,谢谢!
在做标准曲线时是从零即空白开始,做完后在进行标准曲线回归计算时,零点是否一起参加回归,参加与不参加对检测结果有影响吗?
[size=4][url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]是否需要零点校准?如果需要,那么标准曲线法和标准加入法分别用什么溶液校准?[/size]
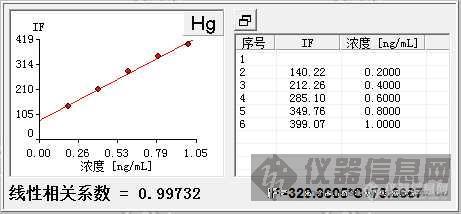
http://ng1.17img.cn/bbsfiles/images/2014/12/201412041652_525914_2903458_3.jpghttp://ng1.17img.cn/bbsfiles/images/2014/12/201412041653_525915_2903458_3.jpg普析原子荧光做汞时曲线拟合零点能达到99.7%,而过零点曲线只有98.4%,标样是现配的,线曲是仪器自动稀释做的,标准空白和标样溶剂都是1%硝酸,为什么会出现这种情况呀,求解。。。

一:相关系数较好名称相关系数斜率截距校准空白试剂空白样品1样品2线性,截距0.9994290.002760.00083-0.30.269 7.5246.659非线性过零点0.9995670.00296000.534 7.1716.338线性过零点0.9988840.00282000.560 7.4186.566二:相关系数不好名称相关系数斜率截距校准空白试剂空白样品1样品2线性,截距0.994360 0.014710.02491-1.6930.592 6.1266.511非线性过零点0.995439 0.02044001.671 4.7575.069线性过零点0.985491 0.01562002.515 5.7716.134http://ng1.17img.cn/bbsfiles/images/2013/05/201305061403_438567_2693958_3.png
sunset有机炭元素碳分析仪怎么零点校准
看很多人好像用纯水或者0.5%HNO3做稀释液,我用的是和最高点一样的作为标准空白和稀释液的,吸光度有0.007,是不过零点的,还有我的样品空白高,所以我现在纠结这个问题,大家都用什么做稀释液,曲线是不是过零点的????







 我要推广仪器
我要推广仪器
 下载APP
下载APP