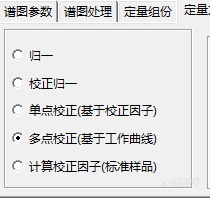
[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]分析方法提供了以下五种方法:[img=,210,198]https://ng1.17img.cn/bbsfiles/images/2022/04/202204241156010412_9653_3138643_3.png!w210x198.jpg[/img]其中归一法和校正归一法有什么区别?是不是归一法不用校正因子,而校正归一法要用校正因子?但是要求校正因子就得做用标准物质算,上面求出的校正因子是绝对校正因子还是相对校正因子?算出校正因子,用归一法和多点校正(基于工作曲线)两种方法计算结果,相差比较大,不知道是什么原因
面积归一化法在色谱分析中有哪些应用?
各位信息网的大侠们,你们谁会安捷伦气相色谱7890B的面积归一化法操作,今天在做正己烷验收测试,要用到面积归一化法,小弟不会啊,还望各位大侠不吝赐教,最好能分享相关文档。谢谢大家。
用面积归一化法走出来的样品结果重现性不好,如何优化色谱条件?
色谱分析定量方法为归一法,分析结果如何以算术平均值报出?
本人是新手很多对液相的知识不懂液相色谱面积归一化法是什么意思啊?具体应该怎么做,求含量?
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]相归一化法计算质量分数的影响因素?谢谢!
气相色谱中的面积百分比法,与归一化法,有何区别?
[color=#444444]本人在做职业卫生方法验证,在做[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法试验中,发现低浓度样品代入标准曲线,得出结果与理论值相差几倍,如果标准曲线强制归零后,结果正常,请问各位高手,[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法回归曲线可以强制归零吗?多谢了!(我们的仪器是岛津GC2030)[/color]
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]可以用面积归一化法来定量吗?
[color=#444444]最近看了一篇文献,是液体a和液体b反应得到液体c的问题,文献上说用“取反应液用 9790 型[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]分析(OV-101[/color][color=#444444]毛细管柱,氢火焰离子化检测器)测定,用峰面积归一法确定产率。“[/color][color=#444444]请问,是怎么计算产率的,能不能提供点知识?[/color]
[table=100%][tr][td]1.[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]归一化法不需要标准物吗?同一样品跑出的峰面积却不同,是怎么回事?2.关于载气流量,我用的是氮气,仪器上都是压力表,方法上都是载气流速如30ml/min如何换算啊,才知道我的压力是多少才能满足条件3.极性毛细管柱可不可以测量非极性的物质,非极性毛细管柱可不可以测量极性物质。说一下为什么。4.氢气启普发生器中的液体是加蒸馏水还是氢氧化钾,(厂家说加蒸馏水可是只加蒸馏水溶度不是变化么[/td][/tr][/table]
[color=#444444]用的是很老的TCD的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url],柱子是GDX-101,前段时间气体关了,但是色谱 没关,在没通气的状态下降温,刚把气体关掉的时候柱温可能在50度左右 ,检测器温度可能在80度左右, 现在用归一法测,测出来的结果就不行了,同一个样,数据能相差10,想问下什么原因,能怎么排除,还有啊,那个校正归一法是不是以归一法为前体,进标样的时候是在归一法下采样的吗?[/color]
谁能教一下[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的面积百分比和归一法的计算
天然生育酚为什么不能用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的面积归一化法来进行检测?
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测花生脂肪酸,归一化法测相对含量。单标混标都要买吗?
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]前段时间峰型不好,出现峰宽增大,峰顶圆头情况,清洗进样口,分流后峰型基本恢复正常,但分析二硝基苯时,同一台仪器同一根色谱柱同一个样品,与未清洗前结果差异较大,见下表,请问这种是什么原因引起的呢?面积归一法[img]https://ng1.17img.cn/bbsfiles/images/2021/11/202111110856234825_4283_1893048_3.png[/img]
谢谢老师的指教,祝新年快乐!还有就是面积归一化法具体是怎么计算的,HP4890色谱仪中的面积法是否等同于归一化法?计算结果是积分仪直接计算出来的吗?还是需要另外换算呢?其相对百分含量能等同于绝对百分含量吗?另外我们公司油脂粗品的酸值有十几之多,也就是说游离脂肪酸很高,可用上法测出的含量没有变化,酸值太高是否不适用该法,我公司没有自动进样器,用外标法恐怕误差太大,用内标的话应选择什么样的内标物较为合适呢?问题太多,谢谢各位的不吝赐教!
跪求!!!人工煤气组分[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法,适用用于便携式的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]吗,
FID色谱,面积归一法,主峰含量比以前低了0.30%,请问在不考虑色谱柱情况下,能否提高主峰含量(0.30%)至以前状态?
色谱出的数据因为要求要按百分含量,在数据进行归一的时候,色谱所出数据有时候只有七八十,但归一后总数有一百对吗?
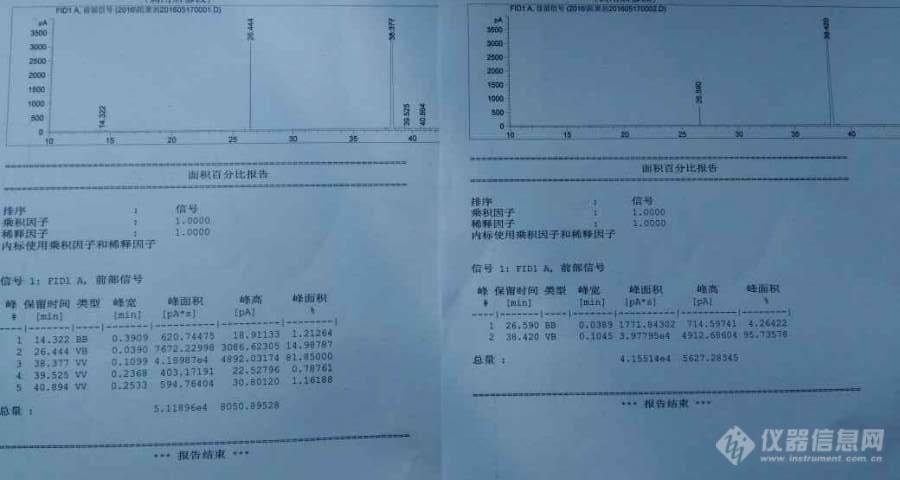
[color=#444444]样品溶剂是乙苯(cas:100-41-4),溶质是4,6二硝基邻仲丁基苯酚(cas:88-85-7),[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]面积归一化法,安捷伦7890,柱箱70℃(10min),10℃/min升至280℃(30min),进样量一微升,分流比30:1。[/color][color=#444444]面积归一化的结果有问题,我最开始做的时候三次结果溶剂和溶质为:1、20%和80,2、40%和60%,3、80%和20%,这结果差异太大了!!!![/color][color=#444444]然后我又找了两个其他的样品,一个是己烷样品一个是工厂的生产后的重组分产品,重复试验,结果两个样品的面积归一化结果都不错,平行的很好。因此我觉得仪器和我的进样技术应该没问题。于是我让同事用分析纯的溶质和溶剂又配了30%的样品,刚才做了两次,结果又差异很大(如下图),第一次是15%和82%,第二次是4%和96%,我真真要疯了!!!!大家能帮我分析分析这是怎么回事吗?[/color][color=#444444][img=,690,368]https://ng1.17img.cn/bbsfiles/images/2019/07/201907091131585211_845_1752342_3.jpg!w690x368.jpg[/img][/color]
我们厂是做液化气分析的,用的是面积归一法,有什么好的办法能一眼就分辨出各种组分的峰!我们做的峰甲烷,乙烷,乙烯,丙烷,丙烯,乙炔,异丁烷,正丁烷,反丁稀,正丁烯,异丁烯,顺丁烯,正戊烷,异戊烷丙炔,1.3丁二烯,但是后面4个峰有时岀峰时间不固定不能准确分辨出是哪个峰
用液相紫外检测器测试标准品(产品)纯度,面积归一方法,取样量多少合适?在应用中样品多一点,色谱柱分离不好,峰型变形,结果偏低,稀释后峰型完美了,结果也高了,平行测定结果偏差较大,哪位在生产岗位应用液相做品控,给提供个最佳取样量?
我们公司是生产淀粉糖的,通常我们做的检测比如:果葡糖浆、葡萄糖粉等产品 都是用的归一化法,原因是简单,能够适应大生产的需要, 最近我们搞实验室认可了 ,,要求用外标法进行检测,这就出问题了,,我们外标法做的数值总比归一化法高2-3% 请问这是方法的问题 还是 仪器 的问题,还是检测人员的问题??? 我们公司用的岛津的液相色谱。
[color=#444444]请问一下,我们实验室用[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]测丙酮中杂质含量,方法是面积归一法,进样量0.2微升,检测结果为0.7562%,分子量为98,如何把他转换成多少ppm的形式啊?请各位大侠明示,感激不尽啊!![/color]
我们用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]面积归一化法测定胺类物质含量,现发现杂质含量偏低(原来正常后突然变低,在其它仪器上测定结果正常),载气压力变大时杂质含量有所增大,不知是何原因,请高手指教!谢谢!
各位,有没有三氯氢硅的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]做法!谢谢啦!
1概述前文简单介绍了载气、管路以及进样口及其各部件可能引起的鬼峰。除此之外,如进样小瓶,隔垫,配样时佩戴的橡胶手套,进样针,洗针液以及色谱柱的流失等亦会导致鬼峰的出现。2[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]峰来源2.1 进样小瓶与瓶盖引入的鬼峰一般地,对于新的未被使用过的进样小瓶与瓶盖是不会引入诸如鬼峰等情况的发生的。进样小瓶引入鬼峰多与瓶盖上所使用的聚合物隔垫有关(单独的进样小瓶,经清洗之后,一般也不会引入鬼峰),且与取样次数,溶解样品所用的溶剂以及样品瓶存放时间有关。进样小瓶盖上的聚合物隔垫,经多次穿透取样,有可能部分聚合物杂质进入样品溶液,甚至与溶剂反生化学反应,在分析图谱上产生一些莫名其妙的鬼峰,影响检测的准确性甚至干扰检测。一般地,进样小瓶盖聚合物隔垫中添加的增塑剂如邻苯二甲酸盐等以及聚合物的分解产物硅氧基烷等均是此类型鬼峰的来源,如下图1所示,为同一个样品小瓶进样多次的对比图。如上图1A所示,新制备的样品的色谱图基线比较干净,在待测物质附近无其他物质干扰,各色谱峰之间分离度较大;在经过数十次重复取样之后,如图1B所示,色谱图上多出了许多的杂峰,已经严重干扰到了对相关物质的检测(包括定性以及定量)。因此,在实际应用时,对于每一个待分析样品,同一进样小瓶最好不要重复多次取样。2.2 橡胶手套以及进样针引入的鬼峰一些偶然性因素如配制样品时所佩戴的橡胶手套,也有可能在接触有机溶剂的时候,样品在转移的过程中被污染而引入鬼峰。如下图2所示,即为两个由橡胶手套引入鬼峰的实例。由进样针引入的鬼峰,多与进样针的类型,洗针液的选择是否合适有关。一般地,洗针液多选择与样品溶液互溶性较好的溶剂(一般为同种溶剂),且需要在使用一定时间之后及时更换。进样针的类型,主要是指针头的形状,一般多选择对进样小瓶盖隔垫以及进样隔垫友好型的进样针,如下图3所示,尽量选择如图3B所示类型针头的进样针,而避免使用如图3A所示类型特别尖锐的针头的进样针。2.3 色谱柱流失及活化,分析物活性引起的鬼峰一般地,新的色谱柱在做一般性物质的分析时是不具有活性的,但不排除由于载气的纯度不够,使用不当等因素,诱发色谱柱产生活性位点,在分析样品时引入鬼峰,如下图4所示。如上图4A所示,在对有关物质进行分析的时候,在样品峰前面出现了特别大的鬼峰,由于其出峰的位置特别靠前,当把入检测器端剪下一段之后,再次对同一样品进行分析的时候,发现鬼峰不见了且基线水平得到降低。色谱柱的入口端以及出口端,一端连接进样口另一端连接检测器,两端的温度均比较高,内部的聚甲基硅氧烷容易被活化形成活性反应位点,或者被样品污染。在使用一段时间之后,需要将两端各切割去一部分,减少色谱峰拖尾甚至鬼峰情况的发生。特别需要注意的是,当使用FID之类的检测器的时候,在载气关闭的时候,最好也将检测器关闭,因为FID检测器在工作的时候,是有水形成的,防止由于反冲进入色谱柱,对色谱柱造成损伤。除了毛细管色谱柱的活化或被污染会导致鬼峰现象之外,色谱柱本身在高温作用下的流失现象也有可能导致鬼峰的产生,且此种现象多与梯度升温程序的设置有关,特别是在完成一个样品的分析之后,色谱柱被迅速降温。由于在迅速降温的过程中,温度分布是不均匀的,色谱柱内部的聚甲基硅氧烷流失成分被滞留在毛细管色谱柱内部,在下一次升温程序启动的时候,随着温度的升高而被带出,表现形式多为有规律性排布的尖刺或者刀阵峰,如下图5所示。对于这种情况,多可以在程序升温的后面编辑程序降温,将色谱柱流失成分充分流出,如下图6所示,获得均匀,连续,稳定的基线。特别是对于一些诸如DB-1701,DB-624类的色谱柱,在对色谱柱进行老化的时候,也建议同上操作。分析物的化学活性,主要是指分析物本身所具有的化学反应性,如与相关溶剂反应,金属接触面接触发生催化反应以及温度不稳定性,如异构化现象(同分异构,扭转异构以及消旋化等)。如下图7所示,分析在梯度升温的过程中,发生了化学转变,而产生了鬼峰现象。一般地,由于分析物本身发生了结构或构象变化或者色谱柱内化学反应而形成的鬼峰,我们可以发现其并不像一个完整意义或者说标准的色谱峰,多伴随有转变平台或者直接就是一个比较钝的三角形或者向下连续延展的平台。下图8A以及8B所示,是两个分析物转化的实例。色谱柱内化学转变,如图8A所示。而对于消旋化或者构象转变,由于并不涉及激烈的化学反应过程,且两种或者几种构象之间在一定的温度下,存在一个较为稳定的平衡态,出现的鬼峰(异构体峰)较多表现为完整意义的色谱峰,且与原分析物色谱峰之间有一定的平台,该平台可通过改变一些仪器参数减小,但基本上不可消除。
最近实验室在做水中的丙酮和苯酚的含量,进过几次样品后,再打纯水也会一直有丙酮和苯酚的峰出来,空白运行时就没有,进样口整棵换成新的,色谱柱也换新的,FID喷嘴也换新的了,结果打纯水进去还是有鬼峰出来。求大神指导







 我要推广仪器
我要推广仪器
 下载APP
下载APP