请问色谱检测时出现平头峰是为什么
请问大家,[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]出平头峰,且平头上还带几个尖尖,是什么原因呀?
最近在做气相色谱6890N测四氯乙烯的标准曲线,原来可以很好的测量,现在在浓度5mg/L时出现平头峰,更高浓度也是平头峰,意思是超出仪器检测限。原来可以测的啊,没有改动,怎么就出现平头峰了?重复配置溶液,还是平头峰。低浓度时峰面积也比正常时大两三倍,什么原因呢?用的自动进样器,定量环为1mL,分流比设为:20:1
使用气相色谱,N3000工作站,面积归一方法时,如果主含量出平头峰对定量有没有影响?请高手解释
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]怎样避免平头峰[em62]
[color=#444444]最近分析异丁烷纯气,为了降低浓度,进入色谱柱时已经用氮气稀释了,PLOT色谱柱,分流比已经调为最大了,但是出峰情况是:稍微放大后看着像是平头峰,这是柱子过载的情况么?因为我用的六通阀定量管是150ul的,没有更小的了,分流比和稀释度也已经最大了,如果过载,是不是只能考虑更换更小的定量管?谢谢[/color][color=#444444][img=,658,424]https://ng1.17img.cn/bbsfiles/images/2019/08/201908021058065098_504_1752329_3.jpg!w658x424.jpg[/img][/color]
气相色谱出平头峰什么原因
[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]分析中,色谱图上有时会看到色谱峰顶点在一段时间内呈现直线,这就是所谓的平头峰。平头峰的出现会造成对组分无法准确定量、定性分析,因此在使用[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]分析时应该尽量避免。 色谱图中出现平头峰,首先要考虑进样量是否过大导致信号过大,信号超过记录仪的zui大测量值,不再上升而出现平头峰,包括进样量过大及饱和度太大等;还可能是检测器灵敏度选择太高,离子化检测器所用静电计输入达到饱和,记录仪滑线电阻或机械部分故障。 一旦遇见平头峰,应该从以下几个方面来解决: 1 减少进样量,或者合理的稀释,加大分流比进样; 2 适当调节检测器信号衰减,改变记录仪量程; 3 增大色谱仪上衰减倍数,减小灵敏度; 当然,在色谱分析时,定量的依据是色谱峰响应大小与组分量在一定范围内呈现线性关系。对有些试验而言,主要考察的是杂质量,所以有时为了能准确检测出有关杂质化合物的量,往往会通过增大供试品溶液浓度,提高杂质的信号响应值来进行试验,这时供试品主峰就可能会出现平头峰,因杂质的响应值与主成分峰的响应值相差太大且无关系,因此不必关注此类主成分平头峰,对杂质自身标准品对照定量法,实际实验中要注意具体问题具体分析
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]出平头峰什么原因?
[align=center][size=24px][b][url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]出现峰面积变大或平头峰等该如何解决[/b][/size][/align][align=left][size=18px] 理想情况下,经[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]分离的峰应该为高斯分布曲线,即对称峰。但实际上当一个样品谱带沿着色谱柱前进时,由于浓度差等原因,样品分子会向谱带两侧扩散,从而使色谱柱出口处的样品谱带比柱入口处宽,且可能产生不对称的峰,就是谱带展宽。 一、不对称峰-谱带展宽 理想情况下,经[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]分离的峰应该为高斯分布曲线,即对称峰。但实际上当一个样品谱带沿着色谱柱前进时,由于浓度差等原因,样品分子会向谱带两侧扩散,从而使色谱柱出口处的样品谱带比柱入口处宽,且可能产生不对称的峰,就是谱带展宽。谱带展宽的程度主要用柱效来表示,色谱峰越对称,峰越窄,柱效越高。 影响谱带展宽的因素有多种,主要分为柱内和柱外两种。柱内因素是指色谱柱本身的性能,如柱活性大小、固定相是否与样品发生化学反应、柱效是否够高、样品是否超载等。柱内因素导致谱带展宽的因素则主要是指街头的死体积、进样口和检测器死体积等。在[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]中,柱外因素导致谱带展宽的程度要比柱内因素小得多。 二、峰面积变大 导致色谱峰峰面积变大的主要原因有以下几点: ①方法参数设置: a.分析条件的改变,如环境[url=http://www.ehsy.com/category-15806]温度[/url]升高,分流比变化等。分流比变小,峰面积变大。 b.数据处理机问题。比如重新开机后,可能会出现这种情况; c.方法设置参数变化,如积分参数变化; d.手动进样时进样技术不好。 ②仪器因素: a.载气流速控制不好,流速增大或柱前[url=http://www.ehsy.com/category-15808]压力[/url]调节阀异常,可能出现这种情况; b.分流口被污染; c.程序升温过程中升温重复性不好,柱温控制不良; ③色谱柱因素: a.色谱柱类型不适合分析该样品,导致固定液流失; b.柱温过高超过了色谱柱固定液的温度上限,导致固定液流失;柱温太靠近色谱固定液的温度下限,导致样品在流动相和固定相之间的分配比发生变化; c.色谱柱用了段时间,未老化,柱性能变差甚至有之前的样品残留物,可能导致这样情况; d.柱温未达到平衡就开始进样; 解决方案如下: 测定时色谱峰面积变大的现象,在确认没有改变色谱条件和方法参数的前提下,首先要考虑进样问题。如果是手动进样,要提高进样技术,进样量要准确、稳定。如果是自动进样,则维护、改善进样系统,保证进样器正常工作; 其次,考察仪器因素。要观察载气压力是否稳定、柱前压调节阀是否有问题;测定分流口和隔垫吹扫口排出的载气,排出气是否减少,必要时调整分流比或清洗分流口。 另外,要记录升温过程(柱温,如有程序升温的进样口也要考察)的温度变化,控温精度是否正常;柱温的控制是否正常尤其重要。 如有问题,需要维修温控系统的电路部分。当然,色谱柱的因素也必须考虑。色谱柱的固定液类型是否适合分离该样品,柱温是否合适等。如果色谱柱不合适,更换色谱柱。 如过色谱柱用了很久,没老化,就老化后再做;如果老化色谱柱,柱性能仍不能恢复,那么得更换新的色谱柱。 三、平头峰 色谱图中出现平头峰,首先要考虑进样量是否过大,导致信号过大,信号超过记录仪的zui大测量值,不再上升出现的平头峰,包括进样量过大及浓度太大等;还可能是检测器灵敏度选择太高,离子化检测器所用静电计输入达到饱和,记录仪滑线电阻或机械部分故障。 解决方案如下: ①减少进样量,或对样品进行合理稀释,或进样时加大分流比; ②适当调节检测器信号衰减,改变记录仪量程; ③增大色谱仪上衰减倍数,减小灵敏度。 当然,在色谱分析时,定量的依据是色谱峰响应大小与组分量在一定范围内呈线性关系。 对有些试验而言,主要考察的是杂质量,所以有时为了能准确检测出有关杂质化合物的量,往往会通过增大供试品溶液浓度,提高杂质的信号响应值来进行试验,这时供试品主峰就可能会出现平头峰,因杂质峰的响应值与主成分峰的响应值相差太大且无关,因此不必关注此类主成分平头峰,对杂质首选自身标准品对照定量法,实际试验中要注意具体问题具体分析。 四、圆头峰 色谱分析中出现圆头峰,有以下几个方面原因: ①进样量过大,超过检测器的线性范围(ECD检测时尤其如此); ②检测器受固定相流失及样品中高沸点成分、易分解组分及腐蚀性物质的污染; ③记录仪灵敏度过低; ④载气系统可能存在泄漏。 解决方案如下: ①减少样品溶液进样量或将样品稀释后再进样,或增大分流比来进样; ②清洗检测器,如果污染物仅限于高沸点物质,则通常可将检测器加热至zui高使用温度后,再通入载气就可清除,要注意加热的温度不能损坏检测器的绝缘材料; 如果加热法不适宜,也可以用丙酮等溶剂从进样口注入(每次可注入几十微升)进行清洗,在污染程度较轻时是有效的;若以上方法都不能解决污染问题,则应将检测器卸下,选择既能溶解污染物又不损坏检测器的溶剂,用注射器注入测量池进行彻底清洗; ③适当调节记录仪灵敏度; ④查看载气气路压力,仔细检查是否存在泄漏,这种情况一般伴随着保留时间或响应值的变化。[/size][/align]
ICP测样时,如果浓度太大过载在谱图上会出现像色谱一样的平头峰或者馒头峰么?
顶空做三氯甲烷四氯化碳,-5的色谱柱,ecd电流0.5nA 四氯化碳最大点10ug/L 三氯甲烷还正常出峰 四氯化碳总是平头峰 信号直接下不来 分流进样 分流比10
[color=#444444]按客户的方法,进的一个纯样品0.2ul,出现平头峰。可是客户那边说:他那也是进的纯的,不出现平头峰。[/color][color=#444444]这个怎么办呢?[/color][color=#444444]是不能加溶剂的。[/color][color=#444444]是色谱柱子的事吗?[/color]
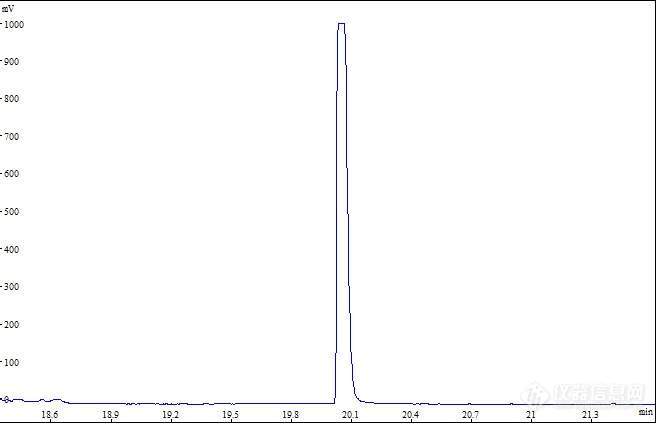
请问出现色谱图平头峰是什么原因?柱子是HP-Innowax,顶空进样,检索离子是m/z44,45等。是柱子问题还是什么问题?前面测定样品使用还正常。[img=,690,517]https://ng1.17img.cn/bbsfiles/images/2023/11/202311211612123014_3161_1615838_3.jpg!w690x517.jpg[/img]
我们有一台瓦里安500型色谱,经过使用一段时间后,出现平头峰.用的是同一标准样,不知为什么色谱图比原来的峰面积大了十几倍左右,从而出现超量程,出现平头峰. 原来的AU值为150mAU,现在用同一样品,进样后,AU值变为1.6个AU,并出现平头峰.不知什么原因, [size=4]请高手赐教!!!!!!!!!!![/size] E-mail: yym3368691@126.com [em53] [em53] [em53] [em53] [em53] [em53] [em53] [em53] [em53] [em53] [em53] [em53] [em53] [em53] [em53] [em53] [em53] [em53] [em53] [em53] [em53] [em53] [em53] [em53]
做样的时候谱图光出平头锋.可以确定不是浓度过高的问题...请求大家帮忙..
用的是FPD硫检测器待测气体是1g煤在微量反应器热解的气体起先用六通阀进样(1ml)出现1400mv的平头峰。后来采用进样针(0.1ml)进样,减小色谱灵敏度,减少反应量1g煤变成0.5g反应,加大反应保护气(稀释气)都会出现平头峰。到底怎么办?跪求方法
问题: 平头峰就是超载吧?含量过高了?亲水性柱子是不是可以走有机相比例很低的流动相?回复: 过载,减少进样量回复2: aq柱可以100%水相回复3: 可以查一下疏水塌陷和亲水作用色谱柱HILIC实用指南回复4: 我正常保持最低5%有机相!不敢再低回复5: 糠醛含量40ppm以下,我记得蜂蜜的旧国标中有,新国标没有看
用气相色谱做克菌丹的农药残留,进标样时大浓度出现平头峰,中小浓度不出峰怎么办?
气相色谱分析中,色谱图上有时会看到色谱峰顶点在一段时间内呈现直线,这就是所谓的平头峰。平头峰的出现会造成对组分无法准确的定性定量分析,因此应尽量避免。
[color=#444444]用佳分的GC9900A [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url],样品已经稀释倍数很大了,浓度已经很小了,为什么主要物质还是会出现平头峰?是该换柱子了么?还有什么其他的原因么,用的是浙大n2000操作系统。[/color]
开机基线无法自动回零点,要用N2000工作站上的零点校正才可回零,进样分析异丙醚样品,纯度98%时在峰高73毫伏时出平头,影响结果呀,但若分析C7混合物,各组分含量都小于25%时,峰高达不到73毫伏,就没平头峰,含量也与其他色谱无差别,请教各位高手指点一下哦。
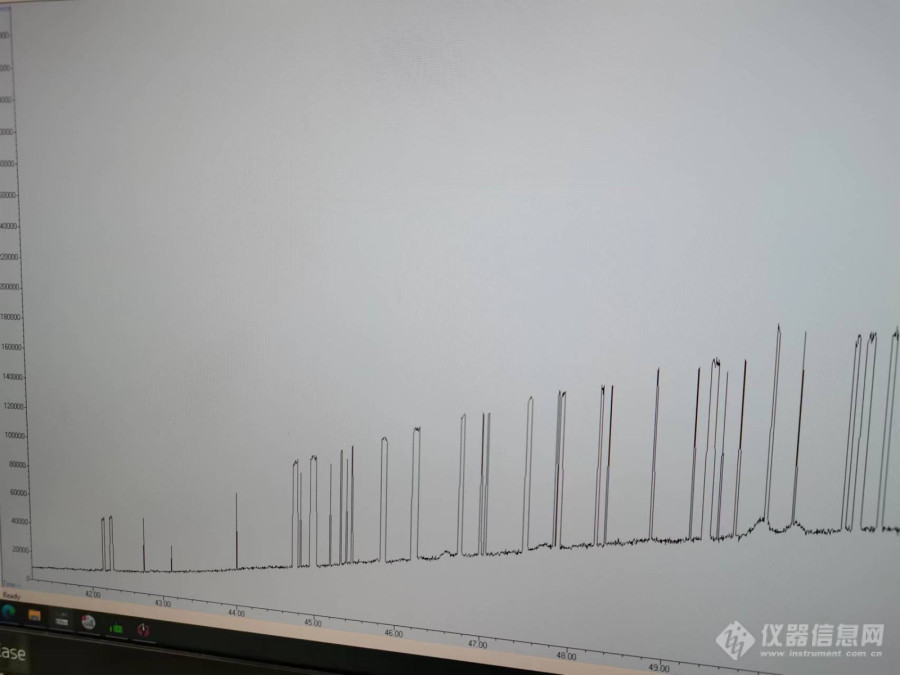
各位老师,我最近检测乙醚和环氧丙烷的时候发现进出来的都是平头峰,更换了色谱柱结果一样。进样浓度4μg/ml,分流比1:1,柱流速1ml/mmin,色谱柱采用HP-INNWAX。[img=,690,401]https://ng1.17img.cn/bbsfiles/images/2019/07/201907221728587791_9417_1853141_3.png!w690x401.jpg[/img]
[color=#444444]用7890[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]做克菌丹的农药残留,大浓度出现平头峰,中小浓度不出峰怎么办?[/color]
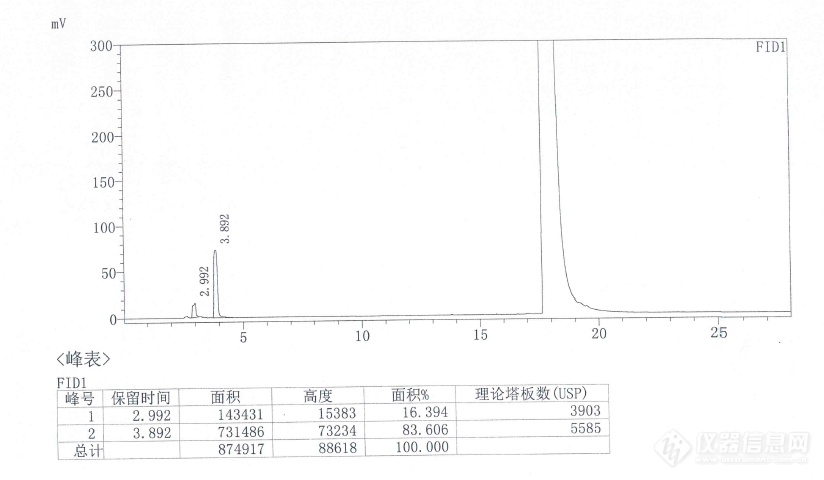
国产的色谱工作站和检测器,峰高就平头,主峰后出现毛刺峰,是哪里的问题???
各位老师,我想请教一下,最近我在打[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的时候,溶剂有平头峰,今天调了一下分流比,进纯溶剂后出峰正常,但在连续进四次溶剂后,溶剂又出现平头峰,这是什么原因?
SP-2000B色谱仪,开机基线无法自动回零点,要用N2000工作站上的零点校正才可回零,分析异丙醚样品,纯度98%峰高73毫伏时出平头,但分析C7混合物含量小于25%,峰高达不到73毫伏,就没有平头
[table=100%][tr][td]别人仪器用的好好的,出峰很正常,我用的时候总是在峰值600处出平头峰,要是浓度太大的话不是应该在仪器最大范围2000出平头峰吗,求解[/td][/tr][/table]
没有满量程,却平头了,而且平头的顶端还是是平线而是类似波浪状两三个峰的比肩,求教老师们这是怎么回事,如何解决呢
是气相色谱存在漏气么?柱子为测试柱OV-101,10m。样品为FID标准测试样,C14,C15,C16溶于异辛烷中。我是菜鸟。谢谢大家。原以为溶剂峰应该为过载平头峰







 我要推广仪器
我要推广仪器
 下载APP
下载APP