推荐厂家
暂无
暂无
 留言咨询
留言咨询
留言咨询
留言咨询
留言咨询
留言咨询

 400-611-9236
留言咨询
400-611-9236
留言咨询
 4006038230
留言咨询
4006038230
留言咨询
 4006038230
留言咨询
4006038230
留言咨询





请问,安捷伦的固相微萃取纤维和色谱科的通用吗?谢谢!
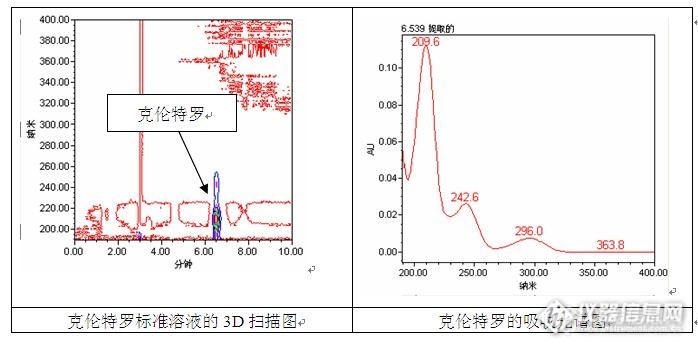
[align=center]固相萃取-高效液相色谱法测定蜂蜜中克伦特罗残留量及条件优化[/align][align=left]1、前言 克伦特罗(Clenbuterol)既不是兽药,也不属于添加剂,而是一种激素类物质,俗称“瘦肉精”,该物质能促进动物体内脂肪分解代谢,增加蛋白质合成,提高瘦肉率,然而人体食用高残留量的内脏组织或者累计摄入量超过一定值时便可能引发食物中毒事件。1997 年我国明令禁止在畜牧行业生产、销售和使用克伦特罗,但是非法使用克伦特罗的事件仍时有发生。 目前,现行有效的标准中测定克伦特罗的方法有酶联免疫法、胶体金免疫层析法、液相色谱法和质谱法,针对不同的样品基质和实验室条件可以选择合适的测定方法。国家标准GB/T 22944-2008中采用液相色谱-串联质谱法测定蜂蜜中克伦特罗的残留量,但是实验室没有[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url],关于高效液相色谱法测定蜂蜜中克伦特罗的文献也不多,于是尝试建立固相萃取-高效液相色谱法测定蜂蜜中克伦特罗的方法并对测试条件进行优化。2、实验方法2.1 仪器和试剂 Waters e2695液相色谱仪(主要包括2998光电二极管矩阵检测器,柱温箱,自动进样器,自动脱气四元梯度泵等);微型漩涡混合仪;12位固相萃取真空装置;12位干浴氮吹仪; Millpore超纯水系统。 甲醇中盐酸克伦特罗标准溶液(250μg/mL,坛墨质检);甲醇、乙酸乙酯和乙酸为色谱纯;乙酸钠、乙酸铵、磷酸二氢钠、氨水和磷酸为分析纯;实验用水为Millpore超纯水系统制得,18MΩ• cm,25 ℃。2.2色谱条件 色谱柱:CNW Athena C18-WP(250mm×4.6mm ,5μm) 流动相:甲醇:磷酸二氢钠(0.02mol/L)=40:60 流速:1.0mL/min 进样体积:20μL 柱温:30℃ 检测波长:210nm3 结果与讨论3.1 检测波长的确定 采用Waters 2998二级管阵列检测器的3D扫描功能,在波长190~400nm范围内对高浓度的克伦特罗标准工作溶液进行测定,并在克伦特罗出峰位置提取光谱图,见下图,从图中可以看出,克伦特罗的最大吸收波长在210nm附近,因此选择210nm作为检测波长,此时克伦特罗的响应值最大。[/align][align=center][img=,690,345]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301354_01_1669358_3.jpg[/img][/align][align=left]3.2 流动相的选择3.2.1甲醇-水为流动相3.2.1.1 流动相pH值对测定结果的影响 首先参考GB/T 5009.192-2003第二法中的色谱条件,以甲醇-水为流动相进行测试,然而结果并不理想,克伦特罗标准溶液在此流动相下并未出峰,几番尝试后决定更换流动相。看到几个采用质谱测定的标准方法均在流动相中加入了酸,于是仍然以甲醇-水为流动相,往水中加入不同体积的磷酸溶液,考察pH值对克伦特罗测定结果的影响(甲醇:水=30:70),测定结果见下图。[/align][align=center][img=,583,845]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301356_01_1669358_3.jpg[/img][/align][align=left] 从上述测定结果中可以发现,未调pH时色谱图中并没有明显的色谱峰,流动相中加入磷酸后克伦特罗在8min左右出峰;随着磷酸加入量的增加,其出峰时间提前并且峰高与峰面积也随着增加;当进一步提高流动相中磷酸含量时,克伦特罗的出峰时间逐渐延长,但是峰面积保持不变。这可能是由于克伦特罗呈弱酸性,在水溶液中以离子形态存在,降低了在C18色谱柱上的保留行为,而磷酸的加入能够抑制克伦特罗的电离,使其以分子的形态存在,增加克伦特罗在C18色谱柱上的保留,并改善峰形。当pH=3.5时,克伦特罗呈部分解离的状态,因此虽然能够出峰,但是峰面积偏小,而当pH=3.0时,克伦特罗则全部以分子形态存在,峰面积保持不变。随着磷酸加入量的增加,克伦特罗的出峰时间延长,峰展宽变大,峰高变小,方法的灵敏度也随着降低。3.2.1.2 流动相比例对测定结果的影响 流动相中有机相比例对高效液相色谱的分离行为有很大影响,调节流动相比例可以改善待测组分的峰形、出峰时间以及与杂质组分的分离度,因此实验中考察了有机相比例对克伦特罗测定结果的影响(pH=2.8),测定结果见下图。从图中可以看出,提高流动相中甲醇含量对克伦特罗的出峰时间有很大的影响,当流动相中甲醇含量为20%时,克伦特罗的出峰时间为17min左右,而当流动相中甲醇含量为45%时,克伦特罗的出峰时间提前到3min左右,而且与溶剂峰重叠,不利于定性和定量分析。[/align][align=center][img=,578,826]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301359_02_1669358_3.jpg[/img][/align][align=left]3.2.1.3 方法重复性测试 根据上述测试结果,初步决定采用甲醇:水(pH=2.8)=30:70的流动相条件进行测试,然而在测试过程中发现,此流动相条件下克伦特罗的保留时间不稳定,随着进样次数的增加,保留时间不断前移,6针进样后克伦特罗保留时间的相对标准偏差(RSD)值为1.3%。产生保留时间漂移的原因可能有两种(1)色谱柱的性能下降,流动相中加了磷酸,色谱柱平衡所需的时间比较长(这是一根服役了很久的色谱柱);(2)此流动相条件的缓冲能力弱,在线混合以及样品的加入导致流动相的pH值发生变化,从而保留时间不稳定。于是修改流动相条件,pH值保持不变,将流动相中甲醇的比例由30%提高至40%,重新考察方法的重复性,实验结果见下图。实验表明,甲醇:水(pH=2.8)=40:60时方法具有较好的重复性,连续6针进样,克伦特罗保留时间的RSD值为0.1%。[/align][align=center][img=,589,419]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301401_01_1669358_3.jpg[/img][/align][align=left]3.2.2甲醇-磷酸盐为流动相 通过上述实验,初步确立了以甲醇-水为流动相测定克伦特罗的高效液相色谱条件,并对部分实验参数进行了优化,然而上述实验结果均是针对克伦特罗标准工作溶液进行的测定,样品成分单一,没有杂质干扰,因此不用考虑杂质与克伦特罗之间的分离度。色谱条件为甲醇:水(pH=2.8)=40:60时虽然解决了方法重复性的问题,但是从色谱图中可以看到,此时克伦特罗的出峰时间较早,仅需3.5min左右就能出峰,而且出峰时间早于溶剂峰,在实际样品分析中很容易受到杂质峰的影响,此方法是否适合测定蜂蜜中的克伦特罗残留量还需进一步验证。 在对蜂蜜样品测定验证之前,先尝试寻找是否有更合适的流动相。以甲醇-水为流动相进行测试时,磷酸的加入对克伦特罗的出峰影响较大,于是尝试改用甲醇-磷酸盐为流动相进行下一步测试。从流动相pH值、流动相比例和方法重复性3个方面对方法进行考察,磷酸盐选用0.02mol/L的磷酸二氢钠,通过磷酸调节流动相的pH值,实验结果见下图。实验表明,以甲醇-磷酸二氢钠(0.02mol/L)为流动相时,有机相比例对克伦特罗出峰时间也有很大的影响,pH值的影响较小,因此后续的实验中直接采用甲醇-磷酸二氢钠(0.02mol/L)为流动相,未对流动相的pH值进行调节。当甲醇:磷酸二氢钠(0.02mol/L)=40:60时,连续6针进样,克伦特罗保留时间的RSD值为0.1%,测试方法具有较好的重复性,同时,克伦特罗的出峰时间远离溶剂峰,可以减小样品中杂质组分对待测物质测定的干扰。[/align][align=center][img=,597,550]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301403_01_1669358_3.jpg[/img][/align][align=left]3.2.3甲醇-乙酸盐为流动相 在查阅文献的过程中发现,也有老师采用乙酸盐缓冲溶液对克伦特罗进行测定,于是决定试一下效果。同样,从流动相pH值、流动相比例和方法重复性3个方面对方法进行考察,乙酸盐选用0.02mol/L的乙酸铵,通过乙酸调节流动相的pH值,实验结果见下图。实验表明,采用甲醇-乙酸铵(0.02mol/L)为流动相时,有机相比例对克伦特罗出峰时间有很大的影响,pH值的影响较小,方法的重复性较好,但是此时基线噪音较大,峰高变小,影响方法的检出限。[/align][align=center][img=,581,588]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301405_01_1669358_3.jpg[/img][/align][align=left] 通过上述实验,分别以甲醇-水、甲醇-磷酸盐和甲醇-乙酸盐为流动相,建立了高效液相色谱法测定克伦特罗的方法,并对部分实验条件进行了优化,在实验中发现了各自方法的优缺点,哪种方法更适合蜂蜜中克伦特罗的测定,还需用蜂蜜样品进行验证。3.3 样品前处理 蜂蜜中通常不含有克伦特罗,即使有,其残留值也很小,而且蜂蜜为半固态粘稠样品,无法直接进样测定,因此需要对蜂蜜样品进行前处理。前处理过程主要包含提取、富集、净化和浓缩4个步骤,此次实验蜂蜜中克伦特罗的提取方法参考GB/T 22944-2008:称取约2g蜂蜜样品于50mL离心管中,加入20mL乙酸钠缓冲溶液(0.2mol/L,pH=5.0),漩涡混匀,蜂蜜完全溶解后备用。 样品中克伦特罗残留量富集、净化的方法有很多种,本次实验分别参照标准GB/T5009.192-2003、SN/T1924-2007、SN/T1924-2011以及GB/T22944-2008,采用CNWBONDWCX(500mg,6mL)、CNWBOND SCX(500mg,6mL)、CNW Poly-Sery MCX(60mg,3mL)和CNW Poly-SeryHLB(500mg,6mL)固相萃取柱对蜂蜜中的克伦特罗残留量进行富集、净化,其中SN/T1924-2007标准已作废,但是标准中所涉及的富集净化方法也曾出现在上海安谱实验科技股份有限公司(以下简称上海安谱)的技术应用文章中,因此对此方法也做了尝试。同时,采用Oasis MCX(60mg,3mL)和OasisHLB(200mg,6mL)固相萃取柱对蜂蜜中的克伦特罗残留量进行富集、净化,简单比较了不同品牌固相萃取柱在净化效果、回收率等性能方面的差异。[/align][align=center][img=,578,358]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301407_01_1669358_3.jpg[/img][/align][align=left] 固相萃取柱所用的填料不同,活化和净化方法也有所区别,具体实验方法如下: (1)WCX固相萃取柱 依次用5mL乙醇和5mL水活化固相萃取柱,将上述蜂蜜提取液全部转移至活化后的固相萃取柱中,分别用5mL水和5mL 乙醇淋洗固相萃取柱,弃去淋洗液,真空抽干2min,最后用10mL氨水-乙醇溶液(体积比2:98)进行洗脱,收集洗脱液。洗脱液在50℃下氮气吹干,准确加入1mL水溶解残渣,过0.22μm滤膜后,进样测定。 (2)SCX固相萃取柱 依次用5mL甲醇、5mL水和5mL0.03mol/L盐酸溶液活化固相萃取柱,将上述蜂蜜提取液全部转移至活化后的固相萃取柱中,分别用5mL水和5mL甲醇淋洗固相萃取柱,弃去淋洗液,真空抽干2min,最后用10mL氨水-甲醇溶液(体积比5:95)进行洗脱,收集洗脱液。洗脱液在50℃下氮气吹干,准确加入1mL水溶解残渣,过0.22μm滤膜后,进样测定。 (3)MCX固相萃取柱 依次用3mL甲醇、3mL水和3mL 0.1mol/L盐酸溶液活化固相萃取柱,将上述蜂蜜提取液全部转移至活化后的固相萃取柱中,分别用3mL 0.1mol/L盐酸溶液、3mL水和3mL50%甲醇淋洗固相萃取柱,弃去淋洗液,真空抽干2min,最后用5mL氨水-甲醇-乙酸乙酯溶液(体积比5:45:50)进行洗脱,收集洗脱液。洗脱液在50℃下氮气吹干,准确加入1mL水溶解残渣,过0.22μm滤膜后,进样测定。 (4)HLB固相萃取柱 依次用5mL甲醇和5mL水活化固相萃取柱,将上述蜂蜜提取液全部转移至活化后的固相萃取柱中,分别用5mL水和5mL甲醇淋洗固相萃取柱,弃去淋洗液,真空抽干2min,最后用10mL氨水-甲醇溶液(体积比5:95)进行洗脱,收集洗脱液。洗脱液在50℃下氮气吹干,准确加入1mL流动相溶解残渣,过0.22μm滤膜后,进样测定。[/align][align=center][img=,578,190]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301408_01_1669358_3.jpg[/img][/align][align=left]3.4 蜂蜜样品的测定3.4.1甲醇-水为流动相 以甲醇-水(pH=2.8)为流动相,测定固相萃取柱净化后的蜂蜜及蜂蜜加标样品,考察该色谱条件对蜂蜜中克伦特罗残留量测定的影响。 当甲醇:水(pH=2.8)=40:60时,测定结果见下图。从图中可以看出,蜂蜜样品提取后直接进样测定色谱图中,在克伦特罗保留时间处有一个很大的杂质峰,虽然该杂质峰经HLB固相萃取柱净化后消失,但是实际检测时该杂质峰对克伦特罗仍然存在隐患,如果净化不干净,就可能出现假阳性的结果,同时,在该流动相条件下,样品在2~6min内有许多杂质峰,影响克伦特罗与杂质组分之间的分离度。对比HLB与MCX固相萃取柱净化后测定的色谱图,在此色谱条件下,HLB的净化效果要优于MCX固相萃取柱,蜂蜜样品经HLB固相萃取柱净化后杂质峰明显减少,MCX固相萃取柱净化后仍有杂质组分会对克伦特罗的测定产生干扰。[/align][align=center][img=,581,863]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301411_01_1669358_3.jpg[/img][/align][align=left] 改变流动相比例,测定蜂蜜加标样品经MCX固相萃取柱净化后的样品,改善克伦特罗与杂质间的分离度,实验结果见下图。从图中可以看出,当甲醇:水(pH2.8)=30:70时克伦特罗与杂质组分的分离度较差,不能实现基线分离;当甲醇:水(pH2.8)=20:80时,克伦特罗与杂质组分虽然能实现基线分离,而且附近没有杂峰干扰,但是此时克伦特罗的峰高变小,灵敏度变低,不利于低浓度克伦特罗残留量的检查。[/align][align=center][img=,584,295]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301413_02_1669358_3.jpg[/img][/align][align=left]3.4.2甲醇-乙酸铵为流动相 以甲醇-乙酸铵(0.02mol/L)为流动相,测定MCX固相萃取柱净化后的蜂蜜加标样品,考察该色谱条件对蜂蜜中克伦特罗残留量测定的影响,实验结果见下图。从图中可以看出,当流动相为甲醇:乙酸铵(0.02mol/L)=60:40时,克伦特罗的测定受到杂质组分的影响很大,克伦特罗出峰时基线较高,而且与杂质组分不能基线分离;当流动相为甲醇:乙酸铵(0.02mol/L)=40:60时,样品中克伦特罗的灵敏度明显降低。[/align][align=center][img=,586,354]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301414_01_1669358_3.jpg[/img][/align][align=left]3.4.3甲醇-磷酸二氢钠为流动相 以甲醇-磷酸二氢钠(0.02mol/L)为流动相,当甲醇:磷酸二氢钠(0.02mol/L)=40:60时,测定固相萃取柱净化后的蜂蜜及蜂蜜加标样品,考察该色谱条件对蜂蜜中克伦特罗残留量测定的影响,实验结果见下图。从图中可以看出,当甲醇:磷酸二氢钠(0.02mol/L)=40:60时,克伦特罗与样品中的杂质实现基线分离,其保留时间附近的干扰组分较少,灵敏度能够满足残留量检测的要求。同时,在此色谱条件下,蜂蜜加标样品只需经过简单的提取便能直接测定,给高残留值蜂蜜样品的测定提供了便捷的方法(直接提取进样时的加标量远高于采用固相萃取柱净化时的加标量)。结合上述实验,最终本实验选择采用甲醇-磷酸二氢钠(0.02mol/L)为流动相对蜂蜜中克伦特罗残留量进行测定。[/align][align=center][img=,578,594]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301416_01_1669358_3.jpg[/img][/align][align=center][img=,580,634]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301417_01_1669358_3.jpg[/img][/align][align=left] 找到合适的流动相后,在该色谱条件下对固相萃取柱的净化效果进行考察。从上图可以看出,乙酸钠缓冲溶液提取后直接进样的色谱图中虽然也能检出克伦特罗,但是其加标量较大(约为固相萃取柱方法的20倍,该实验主要是为了考察杂质对待测物质的干扰),实际样品中克伦特罗的残留值远小于此次的加标量,因此实际样品测定时需要采用固相萃取柱对克伦特罗残留量进行富集、净化和浓缩。对比不同填料固相萃取柱对蜂蜜加标样品净化后测定的色谱图可以发现,以甲醇-磷酸二氢钠(0.02mol/L)为流动相时,CNWBOND SCX固相萃取柱净化后杂质组分的响应值依然很大,SN/T 1924-2007中采用了C18和SCX固相萃取柱串联的方法进行净化,而本实验仅采用了SCX固相萃取柱进行净化,这可能是导致杂质去除不完全的原因之一;采用其他几种固相萃取柱净化后,色谱图中杂质峰的响应值明显降低,其中采用WCX固相萃取柱净化后的色谱图中杂质少,基线比较平整。对比相同填料不同品牌固相萃取柱净化后的色谱图可以发现,两种品牌的固相萃取柱对杂质去除能力难分伯仲。3.5 标准曲线的绘制 准确吸取1.0mL盐酸克伦特罗标准溶液于25mL容量瓶中,用水稀释成浓度为10.0μg/mL的标准储备溶液。吸取适量体积克伦特罗标准储备溶液,用水稀释配成相应浓度的标准工作溶液,并按上述选定的色谱条件进行测定。以样品峰面积Y(mV)对质量浓度X(μg/mL)作图,得到线性回归方程Y=146893X+311,相关系数R2=0.9991,结果表明:克伦特罗含量在0.10~1.00μg/mL之间时,该方法呈现良好的线性关系。标准曲线见下图。[/align][align=center][img=,581,408]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301418_01_1669358_3.jpg[/img][/align][align=left]3.6 回收率的测定 称取约2g蜂蜜样品,共12份,往每份样品中加入0.50mL浓度为1.0μg/mL的克伦特罗标准工作溶液,样品经乙酸钠缓冲溶液提取后,分别采用上述6种固相萃取柱对样品进行净化,每种固相萃取柱做2平行,并以甲醇-磷酸二氢钠(0.02mol/L)为流动相进行测定,考察固相萃取柱的回收率,实验结果见下表。测定结果表明,6种固相萃取柱均具有较好的回收率,回收率大于85%,其中MCX和HLB固相萃取柱的回收率明显高于其他两种填料的固相萃取柱;实验中所使用的相同填料不同品牌的固相萃取柱回收率结果相近。[/align][align=center][img=,690,126]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301420_02_1669358_3.jpg[/img][/align][align=left]4、小结 (1)本实验建立了固相萃取-高效液相色谱法测定蜂蜜中克伦特罗的方法,分别以甲醇-水、甲醇-乙酸盐和甲醇-磷酸盐为流动相,考察了流动相对测定结果的影响。实验结果表明,以甲醇-水为流动相时,流动相对pH值的缓冲能力较弱,克伦特罗的保留时间发生漂移;以甲醇-乙酸盐为流动相时,基线噪音较大,方法灵敏度低;以甲醇-磷酸盐为流动相,方法重复性好,灵敏度较高。 (2)实验中参考不同的标准方法对蜂蜜样品进行前处理,考察了前处理方法对样品净化和回收率影响,同时比较了固相萃取柱性能和品牌对蜂蜜中克伦特罗残留量测定的影响。实验结果表明,固相萃取柱性能对蜂蜜中克伦特罗残留量的测定结果有很大的影响,4种不同填料的固相萃取柱中,MCX和HLB固相萃取柱的回收率较高,WCX和SCX固相萃取柱的回收率略低。 (3)实验中考察了WCX和SCX固相萃取柱的回收率较低的原因:WCX固相萃取柱在乙醇淋洗的过程中会有部分克伦特罗被淋洗下来,从而回收率降低;采用SCX固相萃取柱净化时,洗脱液的碱性需要足够强,否则洗脱不完全,但是提高洗脱液的碱性后,杂质也会一同被洗脱下来。 (4)通过此次实验可以发现,流动相条件和固相萃取柱的选择对样品测定具有很大的影响,不同的流动相其洗脱能力不一样,截止波长也会有差别,实验中以甲醇-乙酸铵为流动相时基线噪音明显大于其中两种流动相;同样是MCX净化后的样品,以甲醇-磷酸盐为流动相时测定得到的样品色谱图中杂质要少于其他两种流动相。 (5)液相色谱虽然对克伦特罗具有较高的响应,但是与质谱相比,质谱具有更高的响应,因此对于残留量很低的样品,还是需要用质谱进行验证,液相方法可以作为前期的筛查手段,[/align]
1.1 正相色谱 八十年代初,人们使用的正相色谱固定相硅胶和吡啶硫氰酸镍盐的络合物晶体能与芳香族化合物形成包合物用于分离芳香族含氮异构体和胆甾醇晶体。已有人[1]将焦炭吸附剂作为填料和键合硅胶作过比较并研究其热力学机理。具有离子化或非离子化功能团的大孔聚合物也开始应用于液相色谱,这些聚合物在整个酸碱范围内稳定,其中一个缺点是当溶剂改变时,它们会热胀冷缩,但是其主要的分析方面用途,即从水中收集痕量研究化合物是没有问题的,通过适当的处理装入色谱柱,一些物质出现过度的峰带变宽,而另一些则出现尖、窄的峰带和高分离度,在强碱性大孔树脂中120分钟内可分离100 种尿液中紫外吸收物质[2]。有人将各种链长度的碳氢配位体键合到硅胶上,根据链长短效应研究保留值的影响,也有过类似的报道,将C22 键合相以及制备键合相担体各种条件作过比较,其键长度和吸附自由能成线性关系。一般地,碱溶液会破坏硅胶,烷基胺比季铵盐更会腐蚀硅胶填料,故通过加填有5mm 硅胶的短预柱来洗脱碱性溶液。Kataev等[3]用聚三氟苯乙烯涂成的硅胶微粒基质并应用到卤代芳烃、多肽和蛋白质的分离。1993年11月,Majors 讨论了在日本发展迅速的HPLC 聚合物填料。Hosoya 制备了大孔聚合乙烯—对—叔丁基苯甲酸丁酯微球和两种别的聚合物微球的性能以及更多的C18HPLC固定相。1.2 反相色谱 固定相的研究进入九十年代更是热火朝天。这方面的研究报道远远超过流动相的研究,人们不断探索保留过程及相关烷基键结构的复杂性和可能制得的各种新型固定相。Bolok 和其合作者从统计技术研究反相色谱柱的老化过程。Montes 等评价了一种硅氢化物介质的硅氢烯制备烷基键合相,更早时候基质制备可供参考。Moriyama等评价了用2mm硅胶制得的TSK胶Super ODS新型反相色谱柱的特性,他们论述了如何分离手性化合物。 Schmid与合作者论述了含饱和脂肪酸键合相的合成与性质,并且与相应的饱和烃固定相的使用作过比较。Pesek和Matyska合成了两种不同的二醇键合相,包括一种是“可靠的”,另一种通过将7-辛烯-1,2-二醇直接连接到氢化硅基质上。Jino和Nakamura [4] 评价了用氟处理的键合硅胶作为一种反相色谱基质与常规反相色谱基质的选择性不同。Buszewki 等比较了烷基胺和烷基键合相基质用于反相分离,发现前者对分离极性化合物效果显著,但其热稳定性不如后者。Wongyai 用苯丙醇胺键合到硅胶上产生离子交换——反相基质的混合模型,并且评价了酸性系列、中性和碱性物质的分离效果。Friebe 将Calixarene键合到硅胶上研究其作为选择相能够形成类似于环糊精的内包合物。Chriswanto 和伙伴们将聚吡咯相涂在硅胶上作为HPLC 的特征化填料。Ge等合成了聚3-辛环吡咯改性硅胶,评价其作为蛋白质的分离及探讨酸、碱介质的稳定性。Skapo和Simpson则在流动床中制得反相基质,他们总结出在有机溶剂中通过这种途径与常规键合相比较可制得重现性更好的基质。Theinpont论述了在HPLC 柱中已经填装好的硅胶的衍生化过程和通过这种途径制得的色谱柱和常规键合相色谱柱进行了比较。1.3 亲和色谱 日本和欧洲较多研究多孔聚合物在液相色谱中的应用,这些聚合物可用于分离各种芳香族化合物和糖类,高分子填料对保留值有较大影响,依据不同溶剂洗脱次序,可估算出聚合物溶解度参数。它们适用于亲水溶质排阻色谱,如有一种吸附剂TSK-SW 硅胶带有亲水键合相,在较大的流速下能经受高压且有非常高的分离度,尤其适用于蛋白质和腐殖酸的分离。曾有人描述过带羟基、氰基、铵离子及芳基金属醚键合到硅胶上,铵离子键合相主要以氢键结合,通过溶剂作用稳定保留值。1.4 离子交换色谱 离子交换固定相的确研究过不少,如八十年代初流行的TSK型SW硅胶及基于二乙胺乙基(DEAE) 制成的阴离子交换材料用于蛋白质及核酸的优化分离,低聚糖的分离采用Micro-Pak AX-5。XAD 树脂是色谱中使用最基本的固定相,弱酸、弱碱及两性电解质的分离均有人研究过,反离子以扩散双电层形式存在。也有人制备多孔甲基丙烯酸盐离子交换剂,但多数人愿意使用商品化的交换剂。Sevec 和Frechet [5] 制得聚甲基丙烯酸缩水甘油酯共聚二甲基乙烯丙烯酸酯色谱柱 (300×8 mm) 用于蛋白质离子交换的制备色谱。他们能做到一次进样到这种色谱柱分离300mg 的蛋白质混合物。Gawdzrk 和 Matynia制备了一种交联的(p,p’—二羟基苯)—丙烷二环氧甘油醚和二乙烯苯的甲基丙烯酸酯的多孔共聚物作为一种最新的HPLC填料并且评价了其作为正相和反相分离的效果。 Danielson 和其合作者[6] 用一种丙胺基硅基质同Kel-F800 反应制得一种弱阴离子交换填料。1.5 空间排阻色谱 Kato 等曾通过丁醚或三乙醇苯醚与TSK G3000 SW 空间排阻色谱法 (SEC) 结合合成 HIC 固定相材料,当使用盐溶液梯度洗脱时,可达到很高的分离度。一些极性键合相通过分配比调节的排阻色谱分离蛋白质和多肽。Miller 等报道过用醚相结构合成大孔硅胶填料:Si-(CH2)3-O-(CH2CH2O)n-R n=1,2或3 R=Me,Et或n-Bu。用这种填料制得的色谱柱可至少连续使用五个月其保留值不变。 烷基链长度和配体密度能直接影响到保留值与分离度,而使用低配位数密度基质更易再生,不易变性,对分离大多数蛋白质来说,基质微球直径在300? 时具有最强的再生力,直径小到1mm的多孔微粒填入1cm 的色谱柱中对于蛋白质的分离十分有用。另一改进色谱有效途径是使用非多孔微球(1-3mm),柱长约30mm,柱效稳定,当小心控制流动相温度和所有仪器组成时,柱外效应很小,曾有人通过洗脱法在2分钟内分离6种蛋白质的混合物。 Smigol[7] 论述了均相聚合甲基丙烯酸缩水甘油酯共聚乙烯二甲基丙烯酸酯微球用特殊化功能的多孔介质制得二醇或二乙胺相有较大孔隙和十八烷基官能团有较少孔隙,这样制得的一种基质当所有分子接近极性相时,较少的分子进入多孔介质,而限制了大分子进入到十八烷基键合相中。 总之,不论是何种类型的固定相对于手性异构体的拆分以及蛋白质和多肽大分子的分离仍在不断的探索完善之中,从发展的趋势看,寻求各种高分子材料作为新型色谱固定相前景光明








 我要推广仪器
我要推广仪器
 下载APP
下载APP