滴定法是最常用的化学分析方法。在滴定中,只要被测组分、滴定剂或反应生成物中的任何一个在紫外光区有特征吸收,并遵守朗伯-比尔定律,就可通过滴定体系的吸光度的变化来指示滴定过程和滴定终点,这就是紫外分光光度滴定法。分光光度滴定法不仅适用紫外光区,也适用可见光区和红外光区。在滴定后,以滴定剂的用量为横坐标,以滴定体系的吸光度为纵坐标,绘制分光光度滴定曲线。滴定曲线中两条直线的交点就是滴定终点。由滴点终点所示的滴定剂体积和浓度,就可计算样品溶液中待测组分的含量或浓度。 分光光度滴定法准确、灵敏和精密度高(相对误差可0.1%),且有适用范围广和可实现自动滴定等特点。有人用这个做过吗
请问测量总氮滴定法和紫外法哪种更精确?
最近做碘化物,紫外分光的碘化物曲线一直做不出来,没有[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url],无奈只能回归到最原始的常规滴定法。做的时候那个17摄氏度后,CPC加完,根本不显色,放冰箱里接近水温0摄氏度时,加CPC才显色,有点惶恐,只能接着滴定,做完发现国标公式是不是也不对,是不是要除以6啊。大神们,快来帮我看看
电位滴定法与永停滴定法电位滴定法与永停滴定法是容量分析中用以确定终点或选择核对指示剂变色域的方法。选用适当的电极系统可以作氧化还原法、中和法(水溶液或非水溶液)、沉淀法、重氮化法或水分测定法等的终点指示。 电位滴定法选用2支不同的电极。1支为指示电极,其电极电势随溶液中被分析成分的离子浓度的变化而变化;另1支为参比电极,其电极电势固定不变。在到达滴定终点时,因被分析成分的离子浓度急剧变化而引起指示电极的电势突减或突增,此转折点称为突跃点。 永停滴定法采用2支相同的铂电极,当在电极间加一低电压(例如50mV)时,若电极在溶液中极化,则在未到滴定终点前,仅有很小或无电流通过;但当到达终点时,滴定液略有过剩,使电极去极化,溶液中即有电流通过,电流计指针突然偏转,不再回复。反之,若电极由去极化变为极化,则电流计指针从有偏转回到零点,也不再变动。仪器装置 电位滴定可用电位滴定仪、酸度计或电位差计,永停滴定可用永停滴定仪或按图示装置。 电流计的灵敏度除另有规定外,测定水分时用10A/格,重氮化法用10A/格。所用电极可按下表选择。 ────────┬──────────┬────────────── 方 法 │ 电 极 系 统 │ 说 明 ────────┼──────────┼────────────── 水溶液氧化还原法│ 铂-饱和甘汞 │铂电极用加有少量三氯化铁的硝 │ │酸或用铬酸清洁液浸洗 ────────┼──────────┼────────────── 水溶液中和法 │玻璃-饱和甘汞 │ ────────┼──────────┼────────────── 非水溶液中和法 │玻璃-饱和甘汞 │饱和甘汞电极套管内装氯化钾的 │ │饱和无水甲醇溶液。玻璃电极用 │ │过后应即清洗并浸在水中保存 ────────┼──────────┼────────────── 水溶液银量法 │ 银-玻璃 │银电极可用稀硝酸迅速浸洗 ├──────────┼────────────── │银-硝酸钾盐桥-饱和甘│ │汞 │ ────────┼──────────┼────────────── -C≡CH中氢置换法│玻璃-硝酸钾盐桥-饱和│ │甘汞 │ ────────┼──────────┼────────────── 硝酸汞电位滴定法│铂-汞-硫酸亚汞 │铂电极可用10%(g/ml)硫代硫酸 │ │钠溶液浸泡后用水清洗。汞-硫 │ │酸亚汞电极可用稀硝酸浸泡后用 │ │水清洗。────────┼──────────┼──────────────永停法 │铂-铂 │铂电极用加有少量三氯化铁的硝 │ │酸或用铬酸清洁液浸洗 ────────┴──────────┴──────────────滴定法 (1)电位滴定法 将盛有供试品溶液的烧杯置电磁搅拌器上,浸入电极,搅拌,并自滴定管中分次滴加滴定液;开始时可每次加入较多的量,搅拌,记录电位;至将近终点前,则应每次加入少量,搅拌,记录电位;至突跃点已过,仍应继续滴加几次滴定液,并记录电位。 滴定终点的确定 用坐标纸以电位(E)为纵坐标,以滴定液体积(V)为横坐标,绘制E-V曲线,以此曲线的陡然上升或下降部分的中心为滴定终点。或以△E/△V(即相邻两次的电位差和加入滴定液的体积差之比)为纵坐标,以滴定液体积(V)为横坐标,绘制(△E/△V)-V曲线,与△E/△V的极大值对应的体积即为滴定终点。也可采用二阶导数确定终点。根据求得的△E/△V值,计算相邻数值间的差值,即为△E/△V,绘制(△E/△V)-V曲线,曲线过零时的体积即为滴定终点。 如系供指示剂变色域的选择核对,滴定前加入指示剂,观察终点前至终点后的颜色变化,以选定该品种终点时的指示剂颜色。 (2)永停滴定法 用作重氮化法的终点指示时,调节R使加于电极上的电压约为50mV。取供试品适量,精密称定,置烧杯中,除另有规定外,可加水40ml与盐酸溶液(1→2)15ml,而后置电磁搅拌器上,搅拌使溶解,再加溴化钾2g,插入铂-铂电极后,将滴定管的尖端插入液面下约2/3处,用亚硝酸钠滴定液(0.1mol/L或0.05mol/L)迅速滴定,随滴随搅拌,至近终点时,将滴定管的尖端提出液面,用少量水淋洗尖端,洗液并入溶液中,继续缓缓滴定,至电流计指针突然偏转,并不再回复,即为滴定终点。用作水分测定的终点指示时,可调节R使电流计的初始电流为5~10μA,待滴定到电流突增至50~150μA,并持续数分钟不退回,即为滴定终点。
目前一般实验室滴定分析采用的是人工滴定法,它是根据指示剂的颜色变化指示滴定终点,然后目测标准溶液消耗体积,计算分析结果。自动电位滴定法是通过电位的变化,由仪器自动判断终点。为了比较仪器和人工滴定方法的测定结果,我们选用了酸价和过氧化值两个指标,分别用自动电位滴定法和人工滴定法进行样品分析。 1 实验部分 1.1 自动电位滴定法的实验仪器 瑞士万通(METROHM)751GPD自动电位滴定仪 727磁力搅拌滴定台 10ml交换单元 6.0431.100Pt电极 6.0133.100pH玻璃电极 6.0729.100Ag/AgCl参比电极 6.0331.0Pt辅助电极 1.2 人工滴定法 按照GB/T5009.37—1996的方法测定样品中的酸价和过氧化值。 2 实验结果与讨论 2.1 两种滴定方法的测定结果对照 自动电位滴定法和人工滴定法测定植物油的酸价和过氧化值结果无显著性差异,表明自动电位滴定仪测定植物油酸价和过氧化值,与现行的国家卫生标准滴定方法结果相近。 2.2 两种滴定方法的精密度比较 选用酸价值较高的样品,分别用自动电位滴定法和人工滴定法平行测定5次,自动电位滴定法测定的相对标准偏差1.1%,人工滴定法为1.6% 平行测定酸价值较低的样品5次,自动电位滴定法测定的相对偏差为2.1%,而人工滴定法的相对标准偏差高达11.4%,表明自动电位滴定法的精密度优于人工滴定法。综上所述,自动电位滴定法测定结果与国标法无异,精密度达到检验要求。由于自动电位滴定法是根据滴定曲线的一阶导数确定终点,等当点与终点的误差非常小,准确度高,避免了人工滴定法由于要加指示剂可能因加入量、指示终点与等当量间、操作者对颜色判断等的误差 电动定位滴定法无须使用指示剂,故对有色溶液、浑浊度以及没有适合指示剂的溶液均可测定 Metrohm自动电位滴定仪可判断多达9个等当点,可以连续滴定溶液中的多个成分,如连续滴定水样中Ca2+、Mg2+,滴定混合酸。自动电位滴定仪还能对滴定分析的各种测定参数,例如测定日期、仪器型号、滴定用标准溶液的消耗量、滴定曲线作自动记录,并自动计算打印出测定结果作为原始记录保存,减少了分析者原始记录数据处理的工作量和运算差错,提高了实验室间分析结果的可比性,有利于实验室管理,因此适于理化分析实验室用作代替人工操作的分析仪器。
本章教学目的:1、掌握氧化还原滴定法分类及各种方法的反应实质。2、了解氧化还原滴定法和酸碱滴定法的异同点。从而明确严格控制反应测定结果的关键。3、熟练掌握碘量法的分析原理、指示剂变色原理及误差来源。教学重点与难点:氧化还原滴定法中各种不同方法的反应实质。教学内容: 一、方法简介1、氧化还原滴定法的分类:氧化还原滴定法:以氧化还原反应为基础的滴定分析方法。标准溶液:氧化剂——测定还原性物质含量。 还原剂——测定氧化性物质含量。氧化还原滴定法: 高锰酸钾法 重铬酸钾法 碘量法 溴酸钾法氧化还原滴定法的特点及与酸碱滴定法的比较:①酸碱滴定法是离子互换反应,反应历程简单、快速。②氧化还原滴定法是电子转移反应,反应复杂、反应速度快慢不一、受外界条件影响较大。比较结果:氧化还原滴定法需要控制反应条件,使其符合滴定分析的要求2、氧化还原滴定法滴定终点的确定①标准溶液自身做指示剂2MnO4- + 5C2O42-ˉ + 16H+ == 2Mn2+ + 10CO2↑ + 8H2O高锰酸钾为紫色,极稀溶液中呈无色。过量的半滴KMnO4,溶液变粉色。②专属指示剂I2 + 2Na2S2O3 == 2NaI + Na2S4O6稍过量的碘标准溶液与溶液中的淀粉指示剂形成浅蓝色。
电位滴定法和永停滴定法有何联系和区别?两者的使用范围有何不同呢?哪种方法更为实用呢?
什么是滴定法,滴定法准确度高不高? 滴定法是将已知浓度的标准溶液均匀缓慢地滴入试祥溶液中,使标准液与试样溶液中的被测组分进行化学反应。用指示剂或其他电测手段判断反应的终点。根据滴定的标准溶液数量,计算出试样溶液中待测组分的含量。在环境监测中,COD、各种形态的氮、卤素离子、亚硫酸离子、氰基离子等许多项目,都用滴定法测定。有时判断仪器分析值是否准确也常用滴定法校验。滴定法使用的计量器具有容量瓶、移液管、滴定管、量筒等。 滴定法与其他方法相比具有方法简单、器具较少、准确可靠等优点。当试样溶液中被测组分浓度较高时,其准确度较[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]法、分光光度法、[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法等高得多。所以,尽管仪器分析方法比较普遍,仍不能完全取代滴定法。但是,滴定法不适用于微量分析。当被溯组分浓度很低时,许多仪器分析比滴定法灵敏度高。而且,滴定法容易受分析者的操作经验和熟练程度所左右。经验多、较熟练的分析人员所得到的分析结果比经验少、不熟练者准确可靠。因此,在滴定法操作中,一方面要求配制的标准溶液准确无误,另一方面要求滴定时观察的液面与眼睛高度严格保持一致。同时还要注意以下事项: (1)不要加热带有刻度的计量容器; (2)各类计量容器必须充分洗净方可使用; (3)取20mL的溶液时,应当用20mL的移液管取一次,一般不用10mL移液管取两次; (4)不要用嘴把溶液吸入移液管,要用吸耳球。
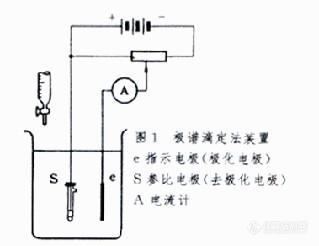
利用电解池中电流的变化指示滴定终点的电滴定分析方法。分为一个极化电极的安培滴定法和两个极化电极的安培滴定法。用滴汞电极为极化电极的一个极化电极的安培滴定法称为极谱滴定法。两个极化电极的安培滴定法称为死停终点法或双安培滴定法。 极谱滴定法是基于极谱法的原理在一定外加电压下滴加标准溶液,藉观察滴定过程中扩散电流的改变以确定滴定终点的容量滴定法。极谱滴定法装置就是一台简易极谱仪加一支滴定管( 图1 )。溶液中被测离子于一定的外加电压下在滴汞电极上还原(或氧化),此时由于浓差极化产生扩散电流。随着被测离子与滴定剂反应浓度越来越低,在滴汞电极上还原(或氧化)而产生的扩散电流越来越小,达到滴定终点时,扩散电流降至零。若将滴定剂体积与每加一次滴定剂后相应的电流读数作图,可得一直线。过滴定终点之后再将滴定剂体积与加滴定剂后的相应电流作图,又得一直线。将所得两条直线延长相交,交点所对应的滴定剂体积即为滴定终点。此法的优点是:①适用范围广,可用于沉淀反应、络合反应和氧化还原反应 ;② 适用浓度范围宽 , 测量范围为0.1~10-4摩尔/升(mol/L)。缺点是 :① 选择性差,易受其他物质干扰;②操作麻烦。 双安培滴定法的装置和图1相似, 两个电极都是铂电极,串联一个电流计指示电流。外加电压一般为几十毫伏。当滴定至电流发生突变时,表示滴定终点到达。滴定曲线的形状取决于滴定体系的可逆性程度( 图2 )。双安培滴定法可用于沉淀反应、络合反应和氧化还原反应。在双安培滴定法中,应用较广的是碘滴定法 ,此外 ,在铈量法测定As3+和Sb3+离子 ,银量法测定卤素离子和CN- 离子也常用此法指示终点 , 此外,也常用来指示库仑滴定法的终点。双安培滴定法的特点是装置简单,准确快速。[img]http://ng1.17img.cn/bbsfiles/images/2010/04/201004221500_213989_1604460_3.jpg[/img]
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=94642]电位滴定法与手工滴定法比较[/url]
电位滴定法快速测定糠醛* 刘俊峰 易平贵 金一粟 摘 要 试验用改良硫酸法由稻草(或麦杆)制取糠醛,用电位滴定法测定糠醛蒸馏液中糠醛的含量及产品的纯度,给出了滴定终点的选择依据.方法简便易行,准确度达到工业分析的要求.图2,表3,参5.关键词 糠醛 电位滴定 条件分类号 TQ201THE QUICK METHOD TO DETERMINE FURFURAL BY ELECTROMETRIC TITRATION Liu Junfeng Yi Pinggui Jin Yisu(Dept. of Chemical Eng. of Xiangtan Mining Institute,Xiangtan ,Hunan,China,411201)ABSTRACT The electrometric titration to determine furfural has been discussed. It also studied the determination method ,the principle, the procedure, the cause for selecting the titration end to determine the pure level of the product and the content of furfural in the distillation liquid produced from straw of rice(or straw of wheat) by the modified sulfuric acid catalytic method. The results show that the method is simple and convenient and its accuracy reaches to the industrial analysis standard.2figs.,3tabs.,5refs.Key words electrometric titration, furfural,conditionSynopsis of the author Liu Junfeng, male, born in 1957, M.E., associate professor, original chemical technology and industrial catalyst. 羰基化合物的测定常见的方法主要有羟胺法、亚硫酸氢钠加成法和2,4-二硝基苯腙重量法[1].上述测定方法有的操作繁杂,有的终点褪色迅速,准确度较差.电位滴定法在羟基苯甲醛产品的分析测定中具有简单、快速、干扰较小等优点[2,3].本文讨论了改良硫酸法制取糠醛产品的测试方法,结果表明,用电位滴定法测定馏出液中糠醛的含量和产品的纯度,结果准确、方法简便易行.1 试验1.1 测定原理[4] 先用NaOH部分中和盐酸羟胺,生成的游离羟胺部分与羰基化合物形成肟,剩余的游离羟胺用盐酸标准溶液回滴,由空白和试样溶液滴定之差值计算试样中糠醛的百分含量.反应如下: 1.2 主要仪器、试剂和溶液 1) 主要仪器 自动电位滴定仪(精度pH=0.01),饱和甘汞电极,玻璃指示电极. 2) 氢氧化钠乙醇溶液( CNaOH=0.75 mol/L) 加入42.0g氢氧化钠(质量分数50%)溶液于688mL乙醇(质量分数95%)中. 3) 羟胺溶液 溶解40.0g盐酸羟胺(NH2OH.HCl)于160mL水中,用95%乙醇溶液稀释至1 L,再加入400 mL浓度为0.75 mol/L氢氧化钠溶液,混匀. 4) 盐酸标准溶液(CHCl=0.250 mol/L) 配制与标定按GB6014.2条进行.1.3 测定步骤与结果计算 称取2.0g(准确至0.0002g)试样于干燥的150mL烧杯中,加入10.0mL含量为95%乙醇,溶解后再加入13mL水,准确吸取25.00mL羟胺溶液至试样中,搅拌反应20min.放入玻璃电极和甘汞电极,在已校正好的酸度计上用0.2500mol/L的盐酸标准溶液滴定至溶液pH值为3.55时终止滴定,消耗体积为V1;除不加试样外,以同样的方法测定25.00mL羟胺溶液空白,消耗体积为V0,计算结果如下: 式中:w糠醛,糠醛的质量分数,%;V0,空白试液消耗盐酸标准溶液体积,mL;V1,被测试样滴定消耗盐酸标准溶液体积,mL;C,盐酸标准溶液浓度,mol/L;m,试样质量,g;系数0.096 09为糠醛的毫摩尔质量,g.2 结果与讨论2.1 测定方法的选择 试验结果发现,对于改良硫酸法制取糠醛产物中醛含量的分析,用亚硫酸氢钠法终点褪色迅速;如用羟胺法直接滴定则无明显滴定突跃,使得终点判断相当困难.唯有羟胺法的间接滴定法终点突跃较为明显,但样品本身的颜色对此法干扰较大,对于颜色较深的样品不适用. 如采用仪器检测法测定样品中的糠醛,由于试样中存在的无机离子和水分在[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]FID检测器上无响应,故归一化结果均偏高.因试样(特别是久置后)本身具有颜色,用可见或紫外吸收光谱仪测定均存在不同程度的干扰而造成结果准确度较差.所以最终选择了电位滴定法.2.2 滴定终点的确定 试验了不同量羟胺溶液滴定后终点pH值的变化,结果如图1. 由图1可知终点pH值分别为3.70、3.58、3.50.根据取样量计算,当试样反应完全后,羟胺剩余量大于15 mL,因此选择pH为3.55时作为滴定终点.计算得此条件下的终点误差约为0.02 mL. 盐酸标准溶液体积/mLa.5mL羟胺溶液滴定曲线(终点pHo 3.70)b.15mL羟胺溶液滴定曲线(终点pHo 3.58)c .25mL羟胺溶液滴定曲线(终点pHo 3.50)图1 羟胺溶液滴定曲线Fig.1 Curve of determining hydroxy amine2.3 滴定反应时间的选择 羰基化合成肟反应一般需30min方能完全,少数情况要在70 ℃以上水浴中进行.本法采用搅拌方式以加快反应速度.测定了室温条件下不同反应时间分析结果的稳定性,如图2.由图2可见,在室温搅拌反应20 min即能满足要求. 图2 不同反应时间糖醛测定含量Fig.2 Content of furfural according different reaction time2.4 测定方法的准确度 采用含量为99.00%的糠醛样品进行准确度试验.在所给试验条件下,测得其含量为99.02%,回收率为100.2%.然后用99.00%含量的糠醛配置标准样品,标样含量为55.26%,实测结果为55.13%,回收率99.76%(见表1),相对误差均符合工业分析公差[5]. 表1 分析方法准确度测定结果 Tab.1 Results of accuracy of the analysis method样品含量/%测定值/%平均值/%回收率/%平均误差/%相对误差/%样品含量/%测定值/%平均值/%回收率/%平均误差/%相对误差/%99.0099.1599.2599.1899.2299.20100.20.0350.2055.2655.1655.1255.1155.1399.760.0200.242.5 试样测定 1) 馏出液糠醛含量的测定 按本试验得出的测定条件,对8批蒸出液先用NaOH中和至pH值与空白试液相同,然后进行糠醛含量的测定,结果见表2. 表2 试样平行测定结果 Tab.2 Results of determination of parallel samples样品号测定值/%平均值/%相对偏差/%样品号测定值/%平均值/%相对偏差/%12.18 2.222.200.0251.561.491.530.0321.961.991.980.0261.641.541.590.0531.881.841.860.0271.411.321.370.0441.721.651.690.0381.361.271.320.04 由表2可见,采用本法两次平行测定结果差值不大于0.2%,相对偏差在0.1%以下. 2) 糠醛产品测定 用上述同样的方法,对连续10批产品进行测定,结果如表3. 表3 产品中糠醛含量测定结果 Tab.3 Determining results of furfural content in products 样品号测定值/%平均值/%相对偏差/%样品号测定值/%平均值/%相对偏差/%196.84 96.8096.820.02696.7296.9096.960.06297.3297.4197.370.05799.3699.2099.280.08395.1695.1095.130.03897.0296.9096.960.06498.2198.1298.170.05997.8297.7697.790.03599.1099.0199.060.041097.7697.5897.670.09 由表3可见,采用本法平行测定结果差值、相对偏差均符合工业分析标准. 3 结论 本法操作简便、仪器简单、准确度、精密度均能满足工业分析要求,可用于糠醛产品质量检测和改良硫酸法生产中糠醛蒸馏液醛含量的分析. *湖南省“八五”科技攻关项目(编号:01-942-57)第一作者简介 刘俊峰 男 42岁 硕士 副教授 有机化工与工业催化剂作者单位:湘潭矿业学院化学工程系,湖南湘潭,411201参考文献 1 西北师院等校编.有机分析教程.北京:人民教育出版社,1978.306~309 2 刘俊峰,易平贵,李晓湘.电位滴定法测定间羟基苯甲醛.分析化学,1996,24(9):1111 3 魏玉鹏.电位滴定法测定对羟基苯甲醛.江苏化工,1992,20(2):45~46 4 兰州大学化学系等编.有机微量定量分析.北京:科学出版社,1978.306~309 5 田景君.分析化学.北京:化学工业出版社,1979.113收稿日期:1998-10-25
电位滴定法快速测定糠醛* 刘俊峰 易平贵 金一粟 摘 要 试验用改良硫酸法由稻草(或麦杆)制取糠醛,用电位滴定法测定糠醛蒸馏液中糠醛的含量及产品的纯度,给出了滴定终点的选择依据.方法简便易行,准确度达到工业分析的要求.图2,表3,参5.关键词 糠醛 电位滴定 条件分类号 TQ201THE QUICK METHOD TO DETERMINE FURFURAL BY ELECTROMETRIC TITRATION Liu Junfeng Yi Pinggui Jin Yisu(Dept. of Chemical Eng. of Xiangtan Mining Institute,Xiangtan ,Hunan,China,411201)ABSTRACT The electrometric titration to determine furfural has been discussed. It also studied the determination method ,the principle, the procedure, the cause for selecting the titration end to determine the pure level of the product and the content of furfural in the distillation liquid produced from straw of rice(or straw of wheat) by the modified sulfuric acid catalytic method. The results show that the method is simple and convenient and its accuracy reaches to the industrial analysis standard.2figs.,3tabs.,5refs.Key words electrometric titration, furfural,conditionSynopsis of the author Liu Junfeng, male, born in 1957, M.E., associate professor, original chemical technology and industrial catalyst. 羰基化合物的测定常见的方法主要有羟胺法、亚硫酸氢钠加成法和2,4-二硝基苯腙重量法[1].上述测定方法有的操作繁杂,有的终点褪色迅速,准确度较差.电位滴定法在羟基苯甲醛产品的分析测定中具有简单、快速、干扰较小等优点[2,3].本文讨论了改良硫酸法制取糠醛产品的测试方法,结果表明,用电位滴定法测定馏出液中糠醛的含量和产品的纯度,结果准确、方法简便易行.1 试验1.1 测定原理[4] 先用NaOH部分中和盐酸羟胺,生成的游离羟胺部分与羰基化合物形成肟,剩余的游离羟胺用盐酸标准溶液回滴,由空白和试样溶液滴定之差值计算试样中糠醛的百分含量.反应如下: 1.2 主要仪器、试剂和溶液 1) 主要仪器 自动电位滴定仪(精度pH=0.01),饱和甘汞电极,玻璃指示电极. 2) 氢氧化钠乙醇溶液( CNaOH=0.75 mol/L) 加入42.0g氢氧化钠(质量分数50%)溶液于688mL乙醇(质量分数95%)中. 3) 羟胺溶液 溶解40.0g盐酸羟胺(NH2OH.HCl)于160mL水中,用95%乙醇溶液稀释至1 L,再加入400 mL浓度为0.75 mol/L氢氧化钠溶液,混匀. 4) 盐酸标准溶液(CHCl=0.250 mol/L) 配制与标定按GB6014.2条进行.1.3 测定步骤与结果计算 称取2.0g(准确至0.0002g)试样于干燥的150mL烧杯中,加入10.0mL含量为95%乙醇,溶解后再加入13mL水,准确吸取25.00mL羟胺溶液至试样中,搅拌反应20min.放入玻璃电极和甘汞电极,在已校正好的酸度计上用0.2500mol/L的盐酸标准溶液滴定至溶液pH值为3.55时终止滴定,消耗体积为V1;除不加试样外,以同样的方法测定25.00mL羟胺溶液空白,消耗体积为V0,计算结果如下: 式中:w糠醛,糠醛的质量分数,%;V0,空白试液消耗盐酸标准溶液体积,mL;V1,被测试样滴定消耗盐酸标准溶液体积,mL;C,盐酸标准溶液浓度,mol/L;m,试样质量,g;系数0.096 09为糠醛的毫摩尔质量,g.2 结果与讨论2.1 测定方法的选择 试验结果发现,对于改良硫酸法制取糠醛产物中醛含量的分析,用亚硫酸氢钠法终点褪色迅速;如用羟胺法直接滴定则无明显滴定突跃,使得终点判断相当困难.唯有羟胺法的间接滴定法终点突跃较为明显,但样品本身的颜色对此法干扰较大,对于颜色较深的样品不适用. 如采用仪器检测法测定样品中的糠醛,由于试样中存在的无机离子和水分在[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]FID检测器上无响应,故归一化结果均偏高.因试样(特别是久置后)本身具有颜色,用可见或紫外吸收光谱仪测定均存在不同程度的干扰而造成结果准确度较差.所以最终选择了电位滴定法.2.2 滴定终点的确定 试验了不同量羟胺溶液滴定后终点pH值的变化,结果如图1. 由图1可知终点pH值分别为3.70、3.58、3.50.根据取样量计算,当试样反应完全后,羟胺剩余量大于15 mL,因此选择pH为3.55时作为滴定终点.计算得此条件下的终点误差约为0.02 mL. 盐酸标准溶液体积/mLa.5mL羟胺溶液滴定曲线(终点pHo 3.70)b.15mL羟胺溶液滴定曲线(终点pHo 3.58)c .25mL羟胺溶液滴定曲线(终点pHo 3.50)图1 羟胺溶液滴定曲线Fig.1 Curve of determining hydroxy amine2.3 滴定反应时间的选择 羰基化合成肟反应一般需30min方能完全,少数情况要在70 ℃以上水浴中进行.本法采用搅拌方式以加快反应速度.测定了室温条件下不同反应时间分析结果的稳定性,如图2.由图2可见,在室温搅拌反应20 min即能满足要求. 图2 不同反应时间糖醛测定含量Fig.2 Content of furfural according different reaction time2.4 测定方法的准确度 采用含量为99.00%的糠醛样品进行准确度试验.在所给试验条件下,测得其含量为99.02%,回收率为100.2%.然后用99.00%含量的糠醛配置标准样品,标样含量为55.26%,实测结果为55.13%,回收率99.76%(见表1),相对误差均符合工业分析公差[5]. 表1 分析方法准确度测定结果 Tab.1 Results of accuracy of the analysis method样品含量/%测定值/%平均值/%回收率/%平均误差/%相对误差/%样品含量/%测定值/%平均值/%回收率/%平均误差/%相对误差/%99.0099.1599.2599.1899.2299.20100.20.0350.2055.2655.1655.1255.1155.1399.760.0200.242.5 试样测定 1) 馏出液糠醛含量的测定 按本试验得出的测定条件,对8批蒸出液先用NaOH中和至pH值与空白试液相同,然后进行糠醛含量的测定,结果见表2. 表2 试样平行测定结果 Tab.2 Results of determination of parallel samples样品号测定值/%平均值/%相对偏差/%样品号测定值/%平均值/%相对偏差/%12.18 2.222.200.0251.561.491.530.0321.961.991.980.0261.641.541.590.0531.881.841.860.0271.411.321.370.0441.721.651.690.0381.361.271.320.04 由表2可见,采用本法两次平行测定结果差值不大于0.2%,相对偏差在0.1%以下. 2) 糠醛产品测定 用上述同样的方法,对连续10批产品进行测定,结果如表3. 表3 产品中糠醛含量测定结果 Tab.3 Determining results of furfural content in products 样品号测定值/%平均值/%相对偏差/%样品号测定值/%平均值/%相对偏差/%196.84 96.8096.820.02696.7296.9096.960.06297.3297.4197.370.05799.3699.2099.280.08395.1695.1095.130.03897.0296.9096.960.06498.2198.1298.170.05997.8297.7697.790.03599.1099.0199.060.041097.7697.5897.670.09 由表3可见,采用本法平行测定结果差值、相对偏差均符合工业分析标准. 3 结论 本法操作简便、仪器简单、准确度、精密度均能满足工业分析要求,可用于糠醛产品质量检测和改良硫酸法生产中糠醛蒸馏液醛含量的分析. *湖南省“八五”科技攻关项目(编号:01-942-57)第一作者简介 刘俊峰 男 42岁 硕士 副教授 有机化工与工业催化剂作者单位:湘潭矿业学院化学工程系,湖南湘潭,411201参考文献 1 西北师院等校编.有机分析教程.北京:人民教育出版社,1978.306~309 2 刘俊峰,易平贵,李晓湘.电位滴定法测定间羟基苯甲醛.分析化学,1996,24(9):1111 3 魏玉鹏.电位滴定法测定对羟基苯甲醛.江苏化工,1992,20(2):45~46 4 兰州大学化学系等编.有机微量定量分析.北京:科学出版社,1978.306~309 5 田景君.分析化学.北京:化学工业出版社,1979.113收稿日期:1998-10-25
咨询一下水质铝的电位滴定法,大家用的是哪一款电位滴定仪,用到的电极式“四苯硼酸根离子电极”和“217型双液接参比电极”
沉淀滴定法 : 是 以沉淀反应为基础的一种滴定分析方法。 沉淀滴定法必须满足的条件: 1. S 小,且能定量完成; 2. 反应速度大; 3. 有适当指示剂指示终点; 4. 吸附现象不影响终点观察。 目前,应用较广泛的是: 生成难溶性银盐的沉淀滴定法 银量法 沉淀反应: Ag + +沉淀滴定法:是 以沉淀反应为基础的一种滴定分析方法。 沉淀滴定法必须满足的条件: 1. S 小,且能定量完成; 2. 反应速度大; 3. 有适当指示剂指示终点; 4. 吸附现象不影响终点观察。 目前,应用较广泛的是:生成难溶性银盐的沉淀滴定法——银量法 沉淀反应: Ag+ +X- ⇌ AgX↓ Ag+ + SCN- ⇌ AgSCN ↓应用:Cl-、Br-、I-、Ag+、SCN-以及能定量地产生这些离子的有机化合物。
咨询一下水质铝的电位滴定法,大家用的是哪一款电位滴定仪,用到的电极式“四苯硼酸根离子电极”和“217型双液接参比电极”
氨氮蒸馏中和滴定法,蒸馏完的空白样加混合指示剂后没滴定直接变紫,水样滴定的时候没有经过黄绿色也是直接滴定到淡紫色,做了好多次都是这样,是指示剂的问题吗,大神帮我分析一下。
教学目的:1、掌握沉淀滴定法对反应的要求。2、掌握银量法确定理论终点的方法原理。3、明确分级沉淀及沉淀转化的概念。4、理解测定氯化物的条件。教学重点与难点:莫尔法(铬酸钾作指示剂)作为教学重点。教学内容: 一、方法简介沉淀滴定法(precipitation titration):也称容量分析法(volumetric precipitation method),以沉淀反应为基础的滴定分析方法。用作沉淀滴定的沉淀反应必须满足以下条件:(1)反应速度快,生成沉淀的溶解度小;(2)反应按一定的化学式定量进行;(3)有准确确定理论终点的方法。应用范围:含量在1%以上的卤素化合物和硫氰化物的测定。解释:沉淀反应很多,但能用于沉淀滴定的沉淀反应并不多,因为很多沉淀的组成不恒定,或溶解度较大,或形成过饱和溶液,或达到平衡速度慢,或共沉淀现象严重等。目前比较有实际意义的是生成微溶性银盐的沉淀反应。Ag+ + Cl- = AgCl↓Ag+ + SCN- =AgSCN↓以这类反应为基础的沉淀滴定法称为银量法。主要测定Cl-、Br-、I-、Ag+ 及SCN-等。如有一些沉淀HgS、PbSO4、BaSO4等也可用于沉淀滴定法,但重要性不及银量法。
请问滴定法是采用比色滴定好,还是电极滴定好?
25 、以直接滴定法,测定固体试样中某组合量时,用同一标准溶液滴定,一次在10℃进行,另一次在30℃进行,其它条件相同,测得的结果是:A :与温度无关 B :30℃时较高 C :10℃时较高
滴定法的方法验证,按照hj168标准,检出限的计算是按照附录A1.3滴定法公式计算?还是可以按照A1.1的公式计算?
电位滴定法和人工滴定法二者有什么优缺点?那位老师知道?
我是一家污水处理厂化验室,近期在做氨氮滴定法的时候,严格按照废水四版的规程操作,但总出现蒸馏后的空白一加显色剂就立即变为淡紫色的问题。另外,标样的检测结果也偏低。那位高手能指教一下啊?
请问下各位大神,制药中枸橼酸钠抗凝剂用HPLC法和滴定法如何比较?液相法是测的枸橼酸,而滴定法测的是枸橼酸钠,没有共同的一个标准品来对比?谢谢各位啦
如何用电位滴定法测染发剂中的氧化物?
各位大侠,三氯化钛滴定法使用范围实用性怎样
用于配位滴定法的反应不一定必备的条件是( )。(A)生成的配合物要足够稳定 (B)反应速度要快(C)生成的配合物的组成必须固定 (D)必须以指示剂确定滴定终点
有做氨氮用蒸馏滴定法的吗?用硫酸标准溶液滴定时最后淡紫色怎么来判断?看了资料有说颜色由红紫色(暖色)逐渐转为淡紫色(冷色),求高手指点!
甲醛滴定法为什么不能准确测定含有多种氨基酸的样品中的氨基氮?
甲醛滴定法为什么不能准确测定含有多种氨基酸的样品中的氨基氮?
复杂样品中可能有磷酸根,滴定法该如何测定呢?请高手指点迷津。







 我要推广仪器
我要推广仪器
 下载APP
下载APP