[color=#333333]《药物溶出度仪机械验证指导原则》发布: 为规范仿制药质量和疗效一致性评价工作,国家食品药品监督管理总局组织制定了《药物溶出度仪机械验证指导原则》,现予发布。在仿制药质量和疗效一致性评价工作过程中,各药品生产企业、研究单位和药品检验机构开展体外溶出试验时,应遵照本指导原则 药品检验机构应加强对企业开展溶出度仪机械验证的技术指导 溶出度仪生产商和销售商应优化相应的技术服务。详细内容学路网医学网整理如下:[/color][b][color=#333333]相关推荐[/color][/b][color=#333333]:[/color][url=https://www.xue63.com/jiaoyuershi/2016-3/14591273645077.html][color=#333333]食品药品监管总局办公厅组织制定《药物溶出仪机械验证指导原则(征求意见稿)》[/color][/url][color=#333333] 本指导原则适用于仿制药质量和疗效一致性评价研究工作中,口服固体制剂体外溶出试验(学路网医学网整理《药物溶出度仪机械验证指导原则》详细内容)所用溶出度仪的机械验证。[/color][color=#333333] 一、概述[/color][color=#333333] 本指导原则中的溶出度仪是指《中华人民共和国药典》(2015年版,以下简称《中国药典》)四部通则〈0931〉溶出度与释放度测定法中第一法和第二法的仪器装置。为保证体外溶出试验数据的准确性和重现性,所使用的溶出度仪应满足《中国药典》要求,同时还需满足本指导原则规定的各项技术要求。[/color][color=#333333] 二、验证前检查[/color][color=#333333] 目视检查以下部件:[/color][color=#333333](一)溶出杯[/color][color=#333333] 杯体光滑,无凹陷或凸起,无划痕、裂痕、残渣等缺陷。[/color][color=#333333](二)篮[/color][color=#333333] 篮体无锈蚀,无网眼堵塞或网线伸出,无网眼或篮体变形等现象。[/color][color=#333333](三)篮(桨)轴[/color][color=#333333] 篮(桨)轴无锈蚀,桨面涂层(Teflon或其他涂层)光滑、无脱落。[/color][color=#333333] 三、测量工具[/color][color=#333333] 可采用单一测量工具(如倾角仪、百分表、转速表和温度计等),也可采用模块化的集成测量工具。各种测量工具均应符合相关的计量要求。[/color]
[b]JJF(鄂)42-2018 药物溶出度仪校准规范 电子版!!!!![/b]
各位:有没有人知道光纤药物溶出仪,与普通的溶出仪的相比,优势是什么?谢谢!
[b][color=#444444]药物溶出度简介[/color][/b][color=#444444][/color][color=#444444]指药物从片剂等固体制剂在规定溶剂中溶出的速度和程度。它是评价药物口服固体制剂质量的一个指标,是一种模拟口服固体制剂在胃肠道中崩解和溶出的体外简易试验方法。[/color][color=#444444][/color][color=#333333]仿制药一致性评价、固体制剂新药研发。 [/color][color=#444444][/color][b][color=#444444]药物溶出度仪分类[/color][/b][color=#444444][/color][color=#444444]1.半自动溶出仪和 全自动溶出仪 [/color][color=#444444][/color][color=#444444]2.六杯药物溶出度仪、八杯 药物溶出仪 、十二杯药物溶出仪。[/color][color=#444444][/color][color=#444444][/color][b][color=#444444]药物溶出度仪维护和保养[/color][/b][color=#444444][/color][color=#444444](1)在测定过程中,必须保证水位至少高于水槽右端出水口2cm以上,否则禁止开机。[/color][color=#444444][/color][color=#444444](2)初次开机时,水位应下降1cm。水嘴处有气泡冒出,说明加热箱已有水,否则禁止开机。[/color][color=#444444][/color][color=#444444](3)开机后若无数字显示,电源指示灯不亮,应检查保险丝是否烧断,电源电压是否正常。[/color][color=#444444][/color][color=#444444](4)开机后若温度数字显示乱跳,可查看测浊线插头与传感器插座连接是否良好。[/color][color=#444444][/color][color=#444444](5)不准将温度传感器插入溶出杯溶剂中,以免腐蚀传感器热敏电阻。[/color][color=#444444][/color][color=#444444][/color][b][color=#444444]溶出度仪使用注意事项[/color][/b][color=#444444][/color][color=#444444]按动面板的“复位”键将使计算机复位,即温度窗恢复等待状态,只显示当前水浴实际温度;时钟窗恢复到常规取样时间的预置状态,但显示的是刚才显示的数值;转速窗恢复到转速预置状态,显示的也是刚才显示的数值,运行停止。因此,在正式进行溶出试验以后,请勿随意按动“复位”键!前已述及,也不要随意按动转速窗上的“选择”键!否则转轴将停止转动。[/color][color=#444444][/color][color=#444444][/color][color=#444444]仪器使用完毕后,请把仪器及附件清洗干净,保持清洁[/color][color=#444444].[/color]
[b][color=#444444]药物溶出度简介[/color][/b][color=#444444][/color][color=#444444]指药物从片剂等固体制剂在规定溶剂中溶出的速度和程度。它是评价药物口服固体制剂质量的一个指标,是一种模拟口服固体制剂在胃肠道中崩解和溶出的体外简易试验方法。[/color][color=#444444][/color][color=#333333]仿制药一致性评价、固体制剂新药研发。 [/color][color=#444444][/color][b][color=#444444]药物溶出度仪分类[/color][/b][color=#444444][/color][color=#444444]1.半自动溶出仪和 全自动溶出仪 [/color][color=#444444][/color][color=#444444]2.六杯药物溶出度仪、八杯 药物溶出仪 、十二杯药物溶出仪。[/color][color=#444444][/color][color=#444444][/color][b][color=#444444]药物溶出度仪维护和保养[/color][/b][color=#444444][/color][color=#444444](1)在测定过程中,必须保证水位至少高于水槽右端出水口2cm以上,否则禁止开机。[/color][color=#444444][/color][color=#444444](2)初次开机时,水位应下降1cm。水嘴处有气泡冒出,说明加热箱已有水,否则禁止开机。[/color][color=#444444][/color][color=#444444](3)开机后若无数字显示,电源指示灯不亮,应检查保险丝是否烧断,电源电压是否正常。[/color][color=#444444][/color][color=#444444](4)开机后若温度数字显示乱跳,可查看测浊线插头与传感器插座连接是否良好。[/color][color=#444444][/color][color=#444444](5)不准将温度传感器插入溶出杯溶剂中,以免腐蚀传感器热敏电阻。[/color][color=#444444][/color][color=#444444][/color][b][color=#444444]溶出度仪使用注意事项[/color][/b][color=#444444][/color][color=#444444]按动面板的“复位”键将使计算机复位,即温度窗恢复等待状态,只显示当前水浴实际温度;时钟窗恢复到常规取样时间的预置状态,但显示的是刚才显示的数值;转速窗恢复到转速预置状态,显示的也是刚才显示的数值,运行停止。因此,在正式进行溶出试验以后,请勿随意按动“复位”键!前已述及,也不要随意按动转速窗上的“选择”键!否则转轴将停止转动。[/color][color=#444444][/color][color=#444444][/color][color=#444444]仪器使用完毕后,请把仪器及附件清洗干净,保持清洁[/color][color=#444444].[/color]
我公司生产:生物安全柜,超净工作台,药品阴凉柜,药品冷藏柜,通风柜,空气洁净屏,干燥箱,红外线灭菌器等仪器经销:崩解仪、药物溶出仪、匀浆机、消解仪、均质器、尘埃粒子计数器、酶标仪,洗板机,空气消毒机,低温冰箱,培养箱,等实验室仪器现崩解仪药物溶出仪低价促销,详情QQ3406227086
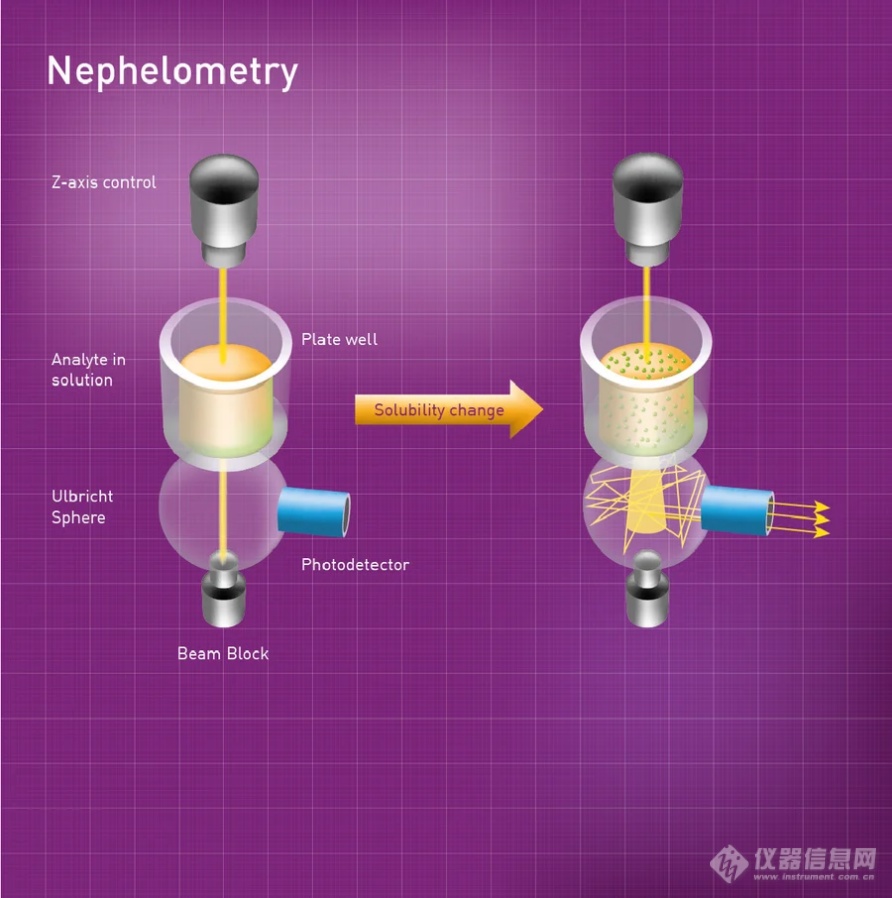
在药物开发过程早期进行ADMET(吸收、分布、代谢、排泄和毒性)评估的能力在当今的药物发现环境中是至关重要的。这意味着需要进行高通量分析,以尽早发现潜在的ADMET问题,从而减少损耗。溶解度是药物的关键特性之一,对分析方法开发、药物生物利用度、吸收和毒性研究,以及药物剂量和药物配方都有重要影响。低溶解度化合物的开发难度更大,获得可再现的ADMET筛选数据也更费时费力。因此,在药物开发的后期阶段进行成本更高的检测之前,研究人员需要一种快速、经济高效的解决方案来确定溶解度。[align=left][b]药物溶解度研究[/b][/align]药物溶解度研究旨在评估药物在不同条件下,在各种溶剂或缓冲液中的溶解度。通常需要测量药物在特定温度或pH值下可溶解的量。溶解度通常表示为药物在溶剂中的最大溶解浓度,也称为饱和浓度。药物溶解度测定在药物发现过程中的不同阶段都至关重要。在早期化学筛选的所有标准中,不理想的溶解度是最不利的性质之一,溶解度低的分子具有很高的失败风险。因此,在药物发现过程中要尽早进行溶解度测定。低溶解度不仅会阻碍新药活性的测试,还可能引发其他不良后果,包括影响其他检测、隐藏其他不良特性,以及对药物动力学和动态性质的潜在影响。总之,这可能会导致药物开发时间大大延迟,或者在尝试改良之前就出现失败。常见的平衡溶解度测定的方法是在恒温条件下将药物和靶标一起振荡至少24小时并测量溶液中的药物浓度(摇瓶法;图 2)。最终浓度通常通过高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法测定,整个过程耗时较长,且通量较低。[b]散射比浊法节省时间并提高通量[/b]散射比浊法是一种快速、可扩展、灵敏且精确的颗粒物质浓度测定方法,有利于药物溶解度研究。另外,这还是一种无损技术,可用于动力学分析,只需制备很少的样品,且可以适应高通量微孔板格式。[url=https://www.bmglabtech.cn/]BMG LABTECH[/url]的NEPHELOstar [i]Plus[/i]是一种专用的微孔板散射浊度仪,可通过测量前向散射光来检测液体样品中的不溶性颗粒。这种方法基于对样品中不溶性颗粒散射光强度的检测。NEPHELOstar [i]Plus[/i]的高强度光源是波长为635 nm的激光。激光穿过样品孔,进入Ulbricht球散射光检测器。如果光线没有被颗粒偏转,会直接穿过球体,不会产生信号。如果样品中存在不溶颗粒,散射光会在球体内部反射,被光电二极管检测到。Ulbricht球可收集散射角度高达 80 度的光线。[align=left][b]结论[/b]散射比浊法是一种快速、可靠、低成本的溶解度筛选方法,可利用384孔板进行高通量筛选。使用NEPHELOstar [i]Plus[/i] 在384孔板中进行全自动动力学溶解度筛选,可在75分钟内分析24种化合物,批间差异率为5%。在提交的化合物中,其中约有90%的化合物,其动力学溶解度可通过此方法得出并排序。[/align]
药物溶出仪的验证方法
各位老大,现在药物溶出仪的验证需要一个验证包,有谁能给详细介绍一下呢?
在人体中,大部分的环境是水相环境,体液、血液和细胞浆液都是水溶液,药物要转运扩散至血液或体液,需要溶解在水中,要求药物有一定的水溶性(又称为亲水性)。而药物在通过各种生物膜(包括细胞膜)时,这些膜是由磷脂所组成的,又需要其具有一定的医`学教育网搜集整理脂溶性(称为亲脂性)。由此可以看出药物亲水性或亲脂性的过高或过低都对药效产生不利的影响。 在药学研究中,评价药物亲水性或亲脂性大小的标准是药物的脂水分配系数,用P来表示,其定义为:药物在生物非水相中物质的量浓度与在水相中物质的量浓度之比。 由于生物非水相中药物的浓度难以测定,通常使用在正辛醇中药物的浓度来代替。Corg表示药物在生物非水相或正辛醇中的浓度;CW表示药物在水中的浓度。P值越大,则药物的脂溶性越高,为了客观反映脂水分配系数的影响,常用其对数lgP来表示。 药物分子结构的改变对药物脂水分配系数的影响比较大。影响药物的水溶医`学教育网搜集整理性因素比较多,当分子中官能团形成氢键的能力和官能团的离子化程度较大时,药物的水溶性会增大。相反若药物结构中含有较大的脂环等非极性结构时,则导致药物的脂溶性增大。 各类药物因其作用不同,对脂溶性有不同的要求。如:作用于中枢神经系统的药医`学教育网搜集整理物,需通过血脑屏障,应具有较大的脂溶性。吸人性的全身麻醉药属于结构非特异性药物,其麻醉活性只与药物的脂水分配系数有关,最适lgP在2左右。
药物溶出仪机械验证指导原则(征求意见稿) 一、概述为进一步推进仿制药与原研药品质量和疗效一致性评价工作的开展,根据《国务院关于改革药品医疗器械审评审批制度的意见》(国发〔2015〕44号)要求,制定本指导原则。在仿制药质量和疗效一致性评价研究工作中,为保证体外溶出试验数据的准确性和重现性,所使用的溶出仪应能够通过本指导原则的各项机械验证技术指标,还应按《中国药典》的要求采用溶出度标准片(如水杨酸片)对仪器进行性能验证试验,均需符合规定。本指导原则适用于仿制药质量和疗效一致性评价体外溶出试验中,《中国药典》2015年版通则0931溶出度与释放度测定法第一法(篮法)和第二法(桨法)所用溶出仪的机械验证。二、机械验证的测量工具溶出仪的机械验证,应将待测部件置于正常溶出试验位置,由相关技术人员使用适宜的测量工具测量各项机械参数。可采用单一测量工具(如倾角仪、同轴度测量工具、摆度表、深度表、转速计和温度计等),也可采用模块化集成测量工具。不论哪种测量工具,均应在检定合格周期内使用,并能够进行量值溯源。三、机械验证的周期溶出仪在安装、移动或维修后都应对其进行机械验证。除另有规定外,通常每六个月对溶出仪进行一次机械验证。如果在试验过程中发现异常现象,应立即对溶出仪进行机械验证。若溶出仪不常使用,可适当延长验证周期,一般不超过12个月。四、机械验证前的检查溶出仪的仪器装置除应符合现版《中国药典》2015年版通则0931溶出度与释放度测定法第一法(篮法)和第二法(桨法)的要求,还应目视检查以下部件:(一)溶出杯。杯体光滑,无凹陷或凸起,无划痕、裂痕、残渣等缺陷。(二)转篮。篮体无锈蚀,无网眼堵塞或网线伸出,无网眼或篮体变形等现象。(三)篮(桨)轴。篮(桨)轴上无锈蚀现象,桨面涂层(Teflon或其他涂层)光滑、无脱落。五、机械验证的流程使用适宜的测量设备,按以下步骤对溶出仪进行机械验证。(一)溶出仪的水平度在溶出杯的水平面板上从两个垂直方向上测量,倾斜度均不得超过0.5°。(二)篮(桨)轴垂直度紧贴篮(桨)轴测量垂直度,再沿篮(桨)轴旋转90°测量,每根篮(桨)轴两次测量数值均不得超过0.5°。(三)溶出杯的垂直度沿溶出杯内壁(避免触及溶出杯底部圆弧部分)测量垂直度,再沿内壁旋转90°测量,每个溶出杯两次测量数值均不得超过1.0°。(四)溶出杯与篮(桨)轴的同轴度篮法:一个测量点位于篮上方距篮上缘2mm,另一个测量点位于篮上方距篮上缘60mm。桨法:一个测量点位于桨叶上方距桨叶上缘2mm,另一个测量点位于桨叶上方距桨叶上缘80mm。在上述两个测量点,每个溶出杯轴心与篮(桨)轴轴心的偏差均不得超过1.0mm。通过了垂直度与同轴度验证的篮轴、桨和溶出杯均应编号,在溶出杯上缘与固定装置相连的位置上做好标记。在进行溶出度试验时,应将各篮轴、桨和溶出杯放在原已通过验证的位置上,保持各溶出杯与固定装置的相对位置不变。为满足同轴度要求,在调整了溶出杯的位置后应重新验证其垂直度。(五)篮(桨)轴的摆动在篮(桨叶)上方20mm处测量。篮(桨)轴以每分钟50转旋转时,连续测量15秒钟,每根篮(桨)轴的摆动不得超过1.0mm。(六)篮的摆动在篮下缘处测量。篮轴以每分钟50转旋转时,连续测量15秒钟,每个篮的摆动不得超过1.0mm。通过了摆动验证的篮应编号,在进行溶出度试验时,应将各篮放在原已通过验证的位置上,保持与固定装置的相对位置不变。(七)篮(桨)的深度测量每个溶出杯内篮(桨)下缘与溶出杯底部的距离,均应为25mm±2mm。(八)篮(桨)轴的转速将篮(桨)轴的转速设定在每分钟50(100)转。连续记录60秒,各篮(桨)轴的转速均应在50(100)±4%转范围内。(九)溶出杯中的温度设定好溶出仪的水浴温度,取水900ml注入各溶出杯中,待温度恒定后,测量各溶出杯内溶出介质的温度,均应为37℃±0.5℃。(十)振动溶出仪运转时,整套装置应保持平稳,溶出仪任一部分(包括所处的环境)不应产生明显的移动或振动(≤0.1mil)。
最近,要组建一个小规模的新实验室,需要重新购置一整套实验室分析仪器,其中就要购买溶出仪。由于资金有限,不考虑进口的,要在国产里面选择。之前也接触过一两家国产溶出仪的产品,想分享我的使用心得,也想大家谈谈自己都使用什么国产溶出仪,效果如何,给我一个采购的参考。() 现在我工作的地方,有3台溶出仪。 其中1台是ZRS-4,原天津大学无线电厂的,应该来说是老字号了。虽说没有什么定时开机、关机等功能,但最基本的控温、转速功能,已经可以满足日常需求了。关键是性能稳定,少说也有十几二十年的历史了,现在还在一线工作,^_^,只是有一段很长时间没有,齿形带老化了,换过一套之外,主要零部件都完好无损。可能唯一的缺点,就是发热量有点大,如果不开空调,开机一段时间,机盖会热得烫手。所以一般不开空调的话,都打开机盖,好散热。曾经做释放度,连续3天满负荷工作,中途关机休息大概1个小时,都能挨过来,出乎我的意料。 另外2台是今年4月份买的,“天津HYD”生产的。才刚买不久就接二连三的出现问题。一个是转桨、转篮等出现锈迹现象,其中一根转桨更加表面爆裂,有黑色锈斑渗出,很是吓人。后来经协商,重新换了经防腐材料包裹的桨篮。虽说我们是做缓控释制剂,释放度一做就是24小时,介质一般是0.1M盐酸,但也不至于这么不耐用,严重怀疑桨篮的不锈钢材质是不是偷工减料,以低级别代替高级别材质,很失望。另外是 转杆转动时声响很大,齿形带磨损严重,后来厂家工程师到场检修更换了,才有好转。除了这个,还有就是电路板故障,两台都出现这种现象,电路板负责控制转速的插头烧糊了,经检查是接触不良造成过热,金属插头很单薄,仅靠一片金属片与金属柱接粗,后来直接将插头焊接在电路板上,就没事了。可见,还是材料没下足,可能是因为价格压得太低,材料就纷纷缩水了。 还有,就是我们的兄弟单位,在我们买新的2台溶出仪的同时,也新购买了2台“天津TDTF”的溶出仪,型号都是ZRS-8G的。用了一段时间,也是转杆异响严重,后经厂家调试后正常。另外,是取样装置很奇怪,不是用垫片,而是用一个类似于积木的装置来定取样针的位置,操作人员都说很不习惯。另外,机头跟水浴槽的间距太小,导致取样操作不方便。感觉上没有这家公司以前仪器那么好用。是不是都因为价格竞争太厉害,厂家都不惜降低产品品质来销售。 上面是我在使用溶出仪的一点体会。想抛砖引玉,请大家也谈谈都在哪些品牌的国产溶出仪,说说使用心得和不足之处。据我了解,除了天大天发,国内还有上海黄海、天津新天光等等都有做药物溶出仪,不知道哪位有用过相关产品,也来说说,给我采购一个参考。
我用的是 油酸 蓖麻油 橄榄油 IPM 请问 怎么测药物在它们中的溶解度
多数抗肿瘤药物因其本身的难溶性而无法实现有效的靶向递送,进而严重影响其在临床方面的应用。紫杉醇(Paclitaxel, PTX)是目前临床上应用较为广泛的难溶性抗肿瘤药物之一,其对肺癌、卵巢癌、乳腺癌等均具有很好的治疗作用。为了解决其难溶问题,现用临床注射制剂(Taxol®)是将其溶解于聚氧乙烯蓖麻油和无水乙醇的混合溶媒后再行给药。然而,该制剂因缺乏靶向性,对其他正常组织产生明显的毒副作用;而且添加的聚氧乙烯蓖麻油在体内降解时会释放组胺,引起严重的过敏反应。因此,开发方便安全的靶向给药系统对PTX的临床应用有重要的研究意义。 近日,中科院过程工程研究所马光辉研究员领导的团队开发出了一种新型的难溶性抗肿瘤药物的纳米靶向给药系统(如图所示)。首先,利用O/W/O复乳液法并结合程序升温法,成功地将PTX以纳米晶形式原位装载于亲水性材料羧化壳聚糖纳米球中,并结合快速膜乳化技术实现了纳米球粒径的均一性。在此基础上,研究人员利用纳米球表面的羧基,引入具有隐形效果的聚乙二醇(PEG)链和靶向肿瘤细胞的RGD肽,最终制得兼具隐形和靶向能力的纳米给药系统。 后续的体外细胞及体内荷瘤小鼠模型实验表明,该制剂能够有效延长药物在体内的循环周期,改善纳米球对肿瘤细胞的亲和能力,提高药物生物利用度。另外,与传统的注射制剂相比,该制剂还具有很低的毒副作用。 上述研究工作已发表在Molecular Pharmceutics(2012, 9, 1736-1747)上,审稿人认为这是一项有趣的工作,方法新颖。该研究工作受到973项目(2009CB930300)和国家自然科学基金(20820102036, 21161160555)的资助。http://www.cas.cn/ky/kyjz/201207/W020120720343496926834.jpg PTX靶向纳米给药系统示意图
液质联用,由于测定药物溶解度的关系,在药物母液中加盐酸调了PH。对柱子、质谱有伤害嘛?知道是流动相肯定是不能用盐酸调PH的,但是,有其他前辈有的说母液加盐酸可用,有的说不可以。也问了维修工程师,说应该影响不大,但是他说具体找应用方面的工程师,今天休息。拜托各位了~
各位同仁,现在最好的药物溶出仪是什么牌子,什么型号的呢?准备FDA认证用的,急急急!
日本科学技术振兴机构村上达也博士研究员和饭岛澄男研究小组负责人,最近成功完成了利用碳纳米管(CNH)作为药物传送系统运载载体的基础实验。利用这一药物传送系统,科学家成功地使抗炎症药物地塞米松吸附在碳纳米管内,从而确认了碳纳米管具有缓慢释放药物成分和缓释后保持药效的特性,此项研究成果可大大加速碳纳米管的药物运载研究与开发。 实验中,科学家首先使用1比1的水与乙醇混合溶剂,在室温液相中使药物地塞米松吸附在碳纳米管中。碳纳米管直径为80至100纳米,具有高亲和性,而地塞米松也是一种易于吸附的物质。碳纳米管氧化后,管端部和侧面会出现孔洞,经过对开孔与未开孔碳纳米管进行对比发现,开孔后的碳纳米管吸附地塞米松的药量比未开孔的高出6倍多。碳纳米管出现孔洞后,每克碳纳米管能够吸附200毫克地塞米松。碳纳米管吸附地塞米松后,经过两周时间才能释放出一半吸附量,证明具有缓释特征。地塞米松在试管中有促进骨形成作用,使用碳纳米管中释放出的地塞米松进行试验发现了这一作用。同时发现,在药物释放后也能保持药效。 碳纳米管具有高纯度和尺寸一致等优点,对人体毒性较小,在结构上表面积大,能携带大量药物。科学技术振兴机构的科学家正着手对抗癌药物传送系统进行试验,不久后将进入动物试验阶段。
对于固体药物,其理化性质通常包括:性状(如外观,颜色,物理状态);熔点或沸点;比旋度,溶解性,吸湿性,溶液pH, 分配系数,解离常数,除此之外,对于将用于制剂生产的药物,还需要了解其物理形态,如多晶型、溶剂化物或水合物等,那么对于多晶型和溶剂化物应该如何进行研究?或者说关于这一部分信息应该如何进行叙述?
抗菌药沙星类抗菌药是光毒反应发生率较高的一组,很多名称中都带有“沙星”两个字,如环丙沙星、氧氟沙星、依诺沙星等等,属于抗菌药的喹诺酮类别。据统计,沙星类抗菌药导致光毒反应发生率为0.1~3%,服用该类药物皮肤经光照以后,常见的症状是皮肤红肿、发热、瘙痒、疱疹等。 除杀星类抗菌药物外,还有四环素类、磺胺类、美满霉素、多西环素、地美环素等抗菌药,也有光毒反应的报道。 抗精神病药如氯丙嗪,在临床上,它是用来治疗精神病、镇吐、低温麻醉及人工冬眠等情况的药物。氯丙嗪为处方药,因此在服药前可找专科医生或药师咨询相关情况,服药后也要做好防护,如戴墨镜及避免皮肤直接暴露于阳光下等。 利尿药如速尿、双氢克尿噻(氢氯噻嗪)等,也就是能增加排尿的药物,也是高血压疾病常用的治疗药物,在水肿、心衰、胸水、腹水的也多用到该类药物,临床上也有其发生光毒反应的报道。 抗组胺药如扑尔敏、苯海拉明等均为第一代康组胺药,常用于抗过敏治疗医|学教育网搜集整理。 解热镇痛药如布洛芬、阿司匹林等,很多人对该类药物都不陌生,在感冒、疼痛、消炎时,都可能会用到,在用药日晒后,若出现皮肤反应时,要加以警惕。 降糖药如格列苯脲、格列吡嗪等,该类药物也是用量较大的药物,它们均为处方药,若在服药后出现光毒反应时,应及时找专科医生咨询或换药,切不可擅自更改或停药。 抗肿瘤药:如长春新碱等,长春新碱常用于治疗急性及慢性白血病、恶性淋巴瘤、小细胞肺癌及乳腺癌等,为静脉注射药物,并不是非处方药。
纳米技术改善难溶性药物吸收前景光明 近日,由中国药学会和美国药学科学家协会主办、沈阳药科大学和辽宁省药学会承办的“第二届亚洲阿登制药技术研讨会暨中国药学会药剂专业委员会2010年学术年会”在沈阳召开,会议主题为“难溶药物的剂型策略”。在为期3天的研讨中,与会专家表示,改善难溶性药物的溶解度,促进药物的吸收,提高药物的生物利用度是药剂学领域亟待攻克的难题,而纳米技术这一助推器有助于加速该难题的解决,我国学者应加强相关研究。 溶解度成为制约瓶颈 药物的溶解性是影响药物生物利用度的重要因素之一,难溶性药物因在水中的溶解度小,难以被机体吸收,导致生物利用度较差。随着组合化学、基因技术、高通量筛选技术等在药物研发中的广泛应用,大量具有活性的候选药物被发现。但是,沈阳药科大学崔福德教授表示,由于存在水溶性差的缺陷,四成左右的侯选药物不能上市而限制了其在临床充分发挥疗效。据估计,全球每年约有650亿美元的药品因生物利用度差而造成治疗费用与疗效比例的严重失调。而实际上,许多难溶性药物有着很强的生物活性,在治疗肿瘤、心血管疾病等领域有着良好疗效。因此,如何提高药物的溶解度和吸收率,成为药剂学研究的热点与难点,迫切需要发展新的制剂技术和剂型来解决这一问题。 崔福德介绍,当前,在药剂学研究中提高难溶性药物的溶解度和溶解速率有多种方法,如加入助溶剂、增溶剂和亲水性介质(适用于液体制剂);制成固体分散体和包含物(适用于固体制剂);制成微粒分散系统(适用于液体和固体制剂);还可以采取减少粒径的措施,比如做成药物的纳米结晶(适用于各种剂型)。 “但是这些方法都有一定的局限性。”中国药学会药剂专业委员会主任委员、北京大学药学院张强教授具体分析说,比如成盐类的方法就只适用于一些难溶性弱酸或弱碱类药物,而不适用于所有分子结构的药物;加入助溶剂和亲水性物质的方法,可供选择的溶剂等也是有限的;增溶剂主要是表面活性剂,毒性问题也限制了其使用;包合物同样存在可供选择的品种较少和毒性问题;固体分散体也有老化现象和需要使用大量赋型剂的缺陷;而费用较高和稳定性问题又限制了微粒化方法的使用。 新技术助力难题解决 解决上述问题,纳米技术的应用优势日益显现:纳米化使药物的粒度大大减小,表面积大大增加,水溶性差的药物在纳米载体中可形成较高的局部浓度;药物的黏附性增强,在吸收部位的滞留时间延长;纳米载药系统可以提高药物的透膜能力和稳定性,也有利于提高药物的生物利用度,特别是对于生物药剂学分类体系(BCS)Ⅱ类(低溶解度、高通透性)和Ⅳ类(低溶解度、低通透性)的药物,这一技术越来越受到国内外一些研究机构、制药公司的青睐。 在药剂学领域,一般将制剂中纳米粒子的尺寸界定在1~1000纳米范围,主要包括纳米载体与纳米药物两个方面。纳米载体是指溶解或分散有药物的各种纳米粒,如纳米乳、聚合物纳米粒(纳米囊或纳米球)、脂质纳米粒等;纳米药物则是指直接将原料药物加工成的纳米粒,实质上是微粉化技术、超细粉技术的发展。 张强介绍,纳米乳/微乳是一种由水、油、表面活性剂和一些复合表面活性剂自组装成的粒径小于100纳米的半透明溶液,其易于制备、相对稳定,而且可使大多数水不溶性药物的生物利用度提高显著。自1943年被报道以来,纳米乳/微乳已经得到了广泛的研究,但上市的产品却不多,1995年诺华公司上市了环孢素A的微乳产品,在临床迅速得以推广。现在上市的同类品种还有雷帕霉素自微乳化给药系统。 纳米粒(纳米球或纳米囊)一般是指由天然或合成的高分子材料制成的、粒径在纳米级的固态胶体微粒,可用于包裹亲水性药物,也可包裹疏水性药物,具有在胃肠道中稳定、药物不易被破坏,以及能够调整药物的理化性质、释放和生物学行为等优点。自1976年Birrenbach等人首先提出了纳米粒和纳米囊的概念后,目前以合成高分子材料为聚合物的纳米粒研究得最为广泛。但张强遗憾地表示:“30多年来,这个研究领域没有取得实质性的突破,无论是口服制剂还是注射制剂都没有产品上市。”而天然聚合物的纳米粒所使用的材料包括壳多糖、白蛋白、右旋糖苷、明胶等,其中以口服壳聚糖纳米粒的研究最为广泛。值得一提的是,白蛋白结合紫杉醇纳米粒注射混悬液2005年上市,成为制剂领域的一个重大突破;但口服给药方面仍没有产品面市。 脂质纳米粒是以生物相容性良好的脂质材料为载体,将药物溶解、包裹于脂质核或是吸附于纳米粒表面的新型载药系统。第一代脂质纳米粒是固体脂质纳米粒(SLN),其性质稳定、制备较简便,具有一定的缓释作用,主要适合于难溶性药物的包裹;随后又发展了第二代纳米结构脂质载体(NCL),解决了第一代脂质纳米粒载药量不佳的问题,稳定性也更好。张强谈到,近年来,对脂质纳米粒的研究也相当广泛,特别是第二代脂质纳米粒自1999年开始研究以来,在外用领域如化妆品领域进展很快,开发程度好于脂质体,但至今还没有用于临床的产品。 在表面活性剂和水等附加剂存在下直接将药物粉碎加工成纳米微粒,可以提高药物的吸收或靶向性,特别适合于大剂量的难溶性药物的口服吸收和注射给药,能增加溶出度,提高生物利用度,增加稳定性。此外,它无需载体材料,只有少量的表面活性剂,安全性更高。此类技术分为纳米混悬剂和纳米结晶制备技术。其中,纳米结晶制备技术发展较快,目前已有5种产品利用这种技术生产并在美国上市,包括惠氏公司的Rapamune(西罗莫司)、默克公司的Emend(阿瑞吡坦)、雅培公司的Tricor(非诺贝特)以及Par公司的Megace ES(甲地孕酮)等。
化学药物残留溶剂研究的技术指导原则一、概述 药物中的残留溶剂系指在原料药或辅料的生产中、以及在制剂制备过程中使用或产生而又未能完全去除的有机溶剂。根据国际化学品安全性纲要,以及美国环境保护机构、世界卫生组织等公布的研究结果,很多有机溶剂对环境、人体都有一定的危害,因此,为保障药物的质量和用药安全,以及保护环境,需要对残留溶剂进行研究和控制。 本指导原则是在参考人用药物注册技术要求国际协调会(International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use,ICH)颁布的残留溶剂研究指导原则,美国药典(the United States Pharmacopoeia,USP)、英国药典(British Pharmacopoeia, BP)、欧洲药典(European Pharmacopoeia,EP)、中国药典(Chinese Pharmacopoeia, ChP)相关内容的基础上,结合我国药物研发的特点,通过分析、研究残留溶剂问题与药物的安全性、有效性及质量可控性之间的内在关系而制定的。本指导原则总结了对残留溶剂问题的一般认识,旨在帮助药物研发者科学合理的进行残留溶剂方面的研究,也为药物评价者提供参考。 考虑到残留溶剂研究涉及的范围比较广泛,本指导原则主要对原料药的残留溶剂问题进行讨论,并以此为基础,探讨和总结药物研究过程中对残留溶剂问题的一般性原则。药物研发者可参考本指导原则对制剂和辅料的残留溶剂问题进行研究。 考虑到药物研究开发的阶段性,本指导原则适用于药物研发的整个过程。 二、基本内容 (一)残留溶剂研究的基本原则 1、确定残留溶剂的研究对象 从理论上讲,药物制备过程中所使用的有机溶剂均有残留的可能,均应进行残留量的研究。但是,药物研发者可以通过对有机溶剂的性质、药物制备工艺等进行分析,提出科学合理的依据,有选择性的对某些溶剂进行残留量研究,这样,既可以合理有效的控制产品质量,又有利于降低药物研究的成本,避免不必要的浪费。因此,药物研发者在进行残留溶剂研究之前,需要首先对药物中可能存在的残留溶剂进行分析,以确定何种溶剂需要进行残留量的检测和控制。 2、确定残留溶剂时需要考虑的问题 原料药中有机残留溶剂与其制备工艺密切相关,同时也需要结合其制剂的临床应用特点来考虑如何对可能残留的溶剂进行研究。 2.1 原料药制备工艺 原料药制备工艺中可能涉及的残留溶剂主要有三种来源:合成原料或反应溶剂、反应副产物、由合成原料或反应溶剂引入。其中作为合成原料或反应溶剂是最常见的残留溶剂来源,本部分主要对此进行讨论。 影响终产物中残留溶剂水平的因素较多,主要有:合成路线的长短,有机溶剂在其中使用的步骤,后续步骤中使用的有机溶剂对之前使用的溶剂的影响,中间体的纯化方法、干燥条件,终产品精制方法和条件等等。 2.1.1 合成路线 由于有机化学反应及后处理工艺的复杂性,对于在得到终产物之前的第几步工艺中使用的溶剂可能在终产物中残留不可能有准确定论。但是,一般来说,后面几步中使用的溶剂的残留可能性较大,因此,对于较长路线的工艺,尤其需要关注后几步所使用的各类溶剂。 2.1.2 后续溶剂的影响 后续使用的溶剂对此前使用溶剂的影响是非常复杂的,取决于各溶剂的性质、后续反应中物料状态以及后续步骤除去溶剂的方法等。 2.1.3 中间体的影响 中间体的处理方法、纯化方法和干燥条件等影响中间体的残留溶剂情况,从而影响终产品的溶剂残留情况。 2.2 制剂及其临床应用特点 控制原料药的残留溶剂,最终目的是控制制剂的残留溶剂,使之符合规定。有时候根据制剂的一些特点,可能对原料药残留溶剂的研究和限度要求进行特殊性的考虑。需要注意,以下所列的因素并不是孤立的,在考虑下列因素时需要注意它们之间的相互影响。 2.2.1 剂型、给药途径 不同制剂发挥疗效的机理不同,对其残留溶剂的要求也可能有所不同。例如注射剂与某些局部使用局部发挥药效的皮肤用制剂相比,残留溶剂的要求就可能相对比较严格。 2.2.2 处方 辅料的残留溶剂也是制剂残留溶剂的组成部分。通过对处方中所使用辅料的残留溶剂水平的了解,可以估算原料药中所能允许存在的残留溶剂水平。 2.2.3 工艺 制剂的制备工艺可能引入新的溶剂,也可能使原料药和辅料中的残留溶剂水平降低。例如素片包衣可能引入新的残留溶剂,干燥工艺可能降低残留溶剂水平等。 2.2.4 适应症 出于治疗一些特殊疾病的考虑,有时候较高水平甚至超出安全值水平的残留溶剂也可能被允许,但需要进行充分的利弊分析。 2.2.5 剂量、用药周期 高剂量、长期用药的制剂,与低剂量、短期用药的制剂相比,对于残留溶剂的要求可能相对严格一些。 3、残留溶剂分类及研究原则 根据有机溶剂对人体及环境可能造成的危害的程度,分为以下四种类型进行研究: 3.1 第一类溶剂及研究原则 第一类溶剂是指人体致癌物、疑为人体致癌物或环境危害物的有机溶剂。因其具有不可接受的毒性或对环境造成公害,在原料药、辅料以及制剂生产中应该避免使用。当根据文献或其他相关资料确定合成路线,涉及到第一类溶剂的使用时,建议重新设计不使用第一类溶剂的合成路线,或者进行替代研究。 由于有机溶剂的选用是合成工艺中比较重要的一点,建议替代研究在工艺研究初期即开始进行,这样,有利于将由于溶剂替换对后续的结构确证、质量研究、稳定性考察的影响降至最低。但替代研究是一项比较复杂、耗时的工作,有时候由于条件、时间等的限制,替代研究工作在临床研究前可能无法充分进行。在严格控制残留溶剂量的前提下,可使药物进入临床研究。在临床研究期间、注册标准试行期间、注册标准转正后,仍可进一步进行替代溶剂的研究工作。 因为溶剂的改变可能导致产品物理化学性质以及质量的改变,因此如发生溶剂的替代,则需要进行溶剂改变前后的产品物理化学性质、质量的对比研究,必要时还需要进行结构对比确证,以说明产品的结构是否发生变化。 如果工艺中不可避免的使用了第一类溶剂,则需要严格控制残留量,无论任何步骤使用,均需进行残留量检测。 3.2 第二类溶剂及研究原则 第二类溶剂是指有非遗传毒性致癌(动物实验)、或可能导致其他不可逆毒性(如神经毒性或致畸性)、或可能具有其他严重的但可逆毒性的有机溶剂。此类溶剂具有一定的毒性,但和第一类溶剂相比毒性较小,建议限制使用,以防止对病人潜在的不良影响。 考虑到第二类溶剂对人体的危害以及所使用的溶剂在终产品中残留的可能性,建议对合成过程中所使用的全部第二类溶剂进行残留量研究,以使药物研发者全面掌握产品质量情况,为最终制定合理可行的质量标准提供数据支持。 3.3 第三类溶剂及研究原则 第三类溶剂是GMP或其他质量要求限制使用,对人体低毒的溶剂。第三类溶剂属于低毒性溶剂,对人体或环境的危害较小,人体可接受的粗略浓度限度为0.5%,因此建议可仅对在终产品精制过程中使用的第三类溶剂进行残留量研究。 3.4 尚无足够毒性资料的溶剂及研究原则 这类溶剂在药物的生产过程中可能会使用,但目前尚无足够的毒理学研究资料。建议药物研发者根据生产工艺和溶剂的特点,必要时进行残留量研究。 随着对这类溶剂毒理学等研究的逐步深入,将根据研究结果对其进行进一步的归类。 (二)研究方法的建立及方法学验证 在确定了需要进行残留量研究的溶剂后,需要通过方法学研究建立合理可行的检测方法。目前,常用的检测方法为气相色谱法(Gas Chromatography,GC),也有其他一些检测方法。 1、研究方法的建立 1.1 气相色谱法(GC法) GC法具有检测灵敏度较高,选择性较好的特点,采用此法所需的样品用量较少,基本可以满足所有残留溶剂测定的要求。采用GC法时,需要结合药物和所要检测的溶剂的性质,通过方法学研究确定合适的检测条件。由于通常要同时检测多种溶剂,为操作的可行性和简便性,建议尽量采用同样的检测条件控制尽量多种类的残留溶剂。 1.1.1 进样方法 GC法包括溶液直接进样和顶空进样两种进样方法。通常情况下,沸点低的溶剂建议采用顶空进样法,沸点高的溶剂可以采用溶液直接进样法,当样品本身对测定有影响时,也建议采用顶空进样法。 1.1.2 供试品溶液和对照品溶液的配制 对于固体原料药,如采用溶液直接进样法,需先用水或合适的溶剂使原料药溶解,以使其中的有机溶剂释放于溶液中,才能被准确测定。如采用顶空进样法,通常以水作溶剂;当药物不溶于水,但可溶于一定浓度的酸或碱液中时,可采用不挥发的酸或碱液为溶剂,但不能使用盐酸溶液或氨水;对于非水溶性药物,可采用合适的溶剂,如N,N—二甲基甲酰胺、二甲基亚砜等为溶剂。 不管采用何种进样法,所选择的溶剂应能够尽量同时溶解样品和待检残留溶剂,所
发布日期20040420化学药品溶出度方法研究 唐素芳溶出度(Dissolution r ate)也称溶出速率,是指在规定的溶剂和条件下,药物从片剂、胶囊剂、颗粒剂等固体制剂中溶出的速度和程度。测定固体制剂溶出度的过程称为溶出度试验(Dissolution test),它是一种模拟口服固体制剂在胃肠道中的崩解和溶出的体外试验方法。药物溶出度检查是评价制剂品质和工艺水平的一种有效手段,可以在一定程度上反映主药的晶型、粒度、处方组成、辅料品种和性质、生产工艺等的差异,也是评价制剂活性成分生物利用度和制剂均匀度 的一种有效标准,能有效区分同一种药物生物利用度的差异,因此是药品质量控制必检项目之一。 一般认为,难溶性 (一般指在水中微溶或不溶) 药物,因制剂处方与生产工艺造成临床疗效不稳定的药物以及治疗量与中毒量相接近的药物(包括易溶性药物),其口服固体制剂质量标准中必须设定溶出度检查项。另外固体制剂的处方筛选及生产工艺流程制订过程中,也需对所开发剂型的溶出度做全面考察。一个可行的溶出度试验法应是在不同时间、地点对同一制剂的溶出度测定或不同的操作者之间的测定都必须达到试验结果具有良好的重现性。为了达到以上目的,必须对溶出度测定试验进行 全面充分的研究。 溶出度研究试验主要包括 以下内容:(1)溶出介质的选择,(2) 溶出介质体积的选择,(3)溶出方法(转篮法与桨法)的选择,(4)转速的选择,(5)溶出度测定方法的验证,(6) 溶出度均一性试验(批内),(7)重现性试验(批间)等。 近来在新药审评中发现,部分研究单位在进行溶出度研究时存在一些问题,主要表现在溶出度研究资料过于简单或溶出度研究内容不够全面。现予以具体分析,希望能对溶出度研究有一定的帮助。 1、溶出介质的选择:通常情况下,溶出介质首选水 ,其次是0.1mol/L盐酸、缓冲液(pH值3~8)、人工胃液或人工肠液;若介质中加适量有机溶剂如异丙醇、乙醇或加分散助溶剂如十二烷基硫酸钠(0.5%以下)等,应有文献依据,并尽量选用低浓度,必要时应做生物利用度考察。通过测定药物在不同介质中的溶出曲线(通常应测定至药物全部溶出)来选择适宜的溶出介质。在一些申报资料中,仅简单地通过比较主药在各介质中的溶解度来选择溶出介质;还有一些品种在采用加有表面活性剂、有机溶剂或采用较高pH值的缓冲液为溶出介质时,没有提供充分的试验数据,难以说明介质选择的合理性。 2、溶出介质的体积选择:溶出介质的体积需使药物符合漏槽条件,一般一个剂量单位以溶剂900ml或1000ml为最普遍,规格较小时也可使用常用体积的1/2~3/4。为了满足某些特殊制剂的要求,中国药典自1995年版起增加了小杯法(即溶出度测定法第三法),小杯法常用体积为100~250ml。一些申报资料中,部分品种特别是规格较小的品种,为满足在溶出量测定时药物浓度的需要,在测定溶出度时,将两粒或数粒片剂或胶囊投入1个溶出杯中,这种溶出度试验法是不可行的。因为此时的溶出度测定已是数粒片剂或胶囊的平均溶出度,并没有客观地反映出每粒片剂或胶囊的溶出情况。通常小剂量药物的药效或毒性一般都较高,采用以上方法是不能保证药品的有效性和安全性的,应提请研究者加以注意。 3、转篮法与桨法的选择:一般情况下,片剂多选择桨法,转篮法多用于胶囊剂或漂浮的制剂,研 究资料应进行两种方法的对比试验,以确定最佳方法。 4、转速的选择:目前,各国药典中收载的溶出度测定方法中的转速,大部分在50~100转/分。转篮法以100转/分为主;桨法以50转/分为主。一般认为桨法50转/分相当于转篮法100转/分。转速的设置与具体品种有关,通常,药物制剂的溶出速度随着转速的增加而增大。转速过快,可能会导致对不同制剂溶出行为的区分能力差,所以不推荐选择过高转速。转速的选择应以能区分不同处方和生产工艺的产品为宜, 如确实需要选择高转速,应进行充分的验证。 5 、溶出度测定方法的验证:方法学验证内容与含量测定基本相同,应进行专属性试验(辅料、胶囊壳的干扰试验)、线性试验、回收率试验、溶液稳定性试验等。应该注意的是,在方法学验证中,试验所用的溶媒应为溶出介质,即应考查辅料、胶囊壳在溶出介质中的干扰,药物在溶出介质中的线性、回收率及稳定性等。在一些申报资料中,或者方法学验证内容不全面,或者虽进行了验证, 但所有试验不是在溶出介质中进行的,使审评人员难以判断溶出度方法的可行性。 6、 取样点和限度的确定:通过溶出度均一性试验( 考察同一批样品的溶出曲线)和重现性试验( 考察至少3批样品的溶出曲线),确定合理的 溶出度测定取样点和限度。为避免多次取样造成的误差,测定溶出曲线时取样点不宜过多,通常为5~6个点,小规格的制剂因采用100~250 ml溶出介质,所以溶出曲线一般可选3~4个时间点。限度应综合考虑溶出曲线拐点和一般性要求。 在新药审评的过程中发现,胶囊壳的干扰试验经常被申报单位忽视。在 USP26、BP2000和中国药典2005年版附录(公示稿)中,对空胶囊的干扰试验均有明确的规定,要求也基本一致。经试验考察,若空胶囊的干扰在2%以下时,可忽略不计;大于2%时,应对溶出度测定结果进行校正;大于25%时,则不能通过校正消除干扰, 溶出试验无效,应重新选择溶出度测定方法。 在溶出度研究资料中,另一个被忽视的问题是滤膜吸附情况的考察。在溶出度测定中,溶出液通常经过滤膜(中国药典规定滤膜孔径不大于0.8μm)滤过以得到澄清的溶液,试验所用滤膜应是惰性的,不能明显吸附溶出液中的有效成分,也不能被溶出介质溶出干扰测定的物质。因此在溶出度研究时,一定要首先考察试验所用滤膜对主成分是否有吸附,通常,吸附量在2%以下时可忽略不计; 超过2%时应考虑选用其他滤过方法或更换适宜的溶出介质。 另外在溶出度研究中还应注意溶出度试验仪的校正、溶出介质的脱气以及在操作中要严格执行SOP,保证实验数据的准确性,以全面正确和客观地反映药物的溶出情况。 以上是我个人对药品溶出度研究的几点认识 ,偏颇之处在所难免,欢迎大家批评指正。类别:审评二部
化学药品溶出度方法研究唐素芳20040420溶出度(Dissolution r ate)也称溶出速率,是指在规定的溶剂和条件下,药物从片剂、胶囊剂、颗粒剂等固体制剂中溶出的速度和程度。测定固体制剂溶出度的过程称为溶出度试验(Dissolution test),它是一种模拟口服固体制剂在胃肠道中的崩解和溶出的体外试验方法。药物溶出度检查是评价制剂品质和工艺水平的一种有效手段,可以在一定程度上反映主药的晶型、粒度、处方组成、辅料品种和性质、生产工艺等的差异,也是评价制剂活性成分生物利用度和制剂均匀度 的一种有效标准,能有效区分同一种药物生物利用度的差异,因此是药品质量控制必检项目之一。 一般认为,难溶性 (一般指在水中微溶或不溶) 药物,因制剂处方与生产工艺造成临床疗效不稳定的药物以及治疗量与中毒量相接近的药物(包括易溶性药物),其口服固体制剂质量标准中必须设定溶出度检查项。另外固体制剂的处方筛选及生产工艺流程制订过程中,也需对所开发剂型的溶出度做全面考察。一个可行的溶出度试验法应是在不同时间、地点对同一制剂的溶出度测定或不同的操作者之间的测定都必须达到试验结果具有良好的重现性。为了达到以上目的,必须对溶出度测定试验进行 全面充分的研究。 溶出度研究试验主要包括 以下内容:(1)溶出介质的选择,(2) 溶出介质体积的选择,(3)溶出方法(转篮法与桨法)的选择,(4)转速的选择,(5)溶出度测定方法的验证,(6) 溶出度均一性试验(批内),(7)重现性试验(批间)等。 近来在新药审评中发现,部分研究单位在进行溶出度研究时存在一些问题,主要表现在溶出度研究资料过于简单或溶出度研究内容不够全面。现予以具体分析,希望能对溶出度研究有一定的帮助。 1、溶出介质的选择:通常情况下,溶出介质首选水 ,其次是0.1mol/L盐酸、缓冲液(pH值3~8)、人工胃液或人工肠液;若介质中加适量有机溶剂如异丙醇、乙醇或加分散助溶剂如十二烷基硫酸钠(0.5%以下)等,应有文献依据,并尽量选用低浓度,必要时应做生物利用度考察。通过测定药物在不同介质中的溶出曲线(通常应测定至药物全部溶出)来选择适宜的溶出介质。在一些申报资料中,仅简单地通过比较主药在各介质中的溶解度来选择溶出介质;还有一些品种在采用加有表面活性剂、有机溶剂或采用较高pH值的缓冲液为溶出介质时,没有提供充分的试验数据,难以说明介质选择的合理性。 2、溶出介质的体积选择:溶出介质的体积需使药物符合漏槽条件,一般一个剂量单位以溶剂900ml或1000ml为最普遍,规格较小时也可使用常用体积的1/2~3/4。为了满足某些特殊制剂的要求,中国药典自1995年版起增加了小杯法(即溶出度测定法第三法),小杯法常用体积为100~250ml。一些申报资料中,部分品种特别是规格较小的品种,为满足在溶出量测定时药物浓度的需要,在测定溶出度时,将两粒或数粒片剂或胶囊投入1个溶出杯中,这种溶出度试验法是不可行的。因为此时的溶出度测定已是数粒片剂或胶囊的平均溶出度,并没有客观地反映出每粒片剂或胶囊的溶出情况。通常小剂量药物的药效或毒性一般都较高,采用以上方法是不能保证药品的有效性和安全性的,应提请研究者加以注意。 3、转篮法与桨法的选择:一般情况下,片剂多选择桨法,转篮法多用于胶囊剂或漂浮的制剂,研 究资料应进行两种方法的对比试验,以确定最佳方法。 4、转速的选择:目前,各国药典中收载的溶出度测定方法中的转速,大部分在50~100转/分。转篮法以100转/分为主;桨法以50转/分为主。一般认为桨法50转/分相当于转篮法100转/分。转速的设置与具体品种有关,通常,药物制剂的溶出速度随着转速的增加而增大。转速过快,可能会导致对不同制剂溶出行为的区分能力差,所以不推荐选择过高转速。转速的选择应以能区分不同处方和生产工艺的产品为宜, 如确实需要选择高转速,应进行充分的验证。 5 、溶出度测定方法的验证:方法学验证内容与含量测定基本相同,应进行专属性试验(辅料、胶囊壳的干扰试验)、线性试验、回收率试验、溶液稳定性试验等。应该注意的是,在方法学验证中,试验所用的溶媒应为溶出介质,即应考查辅料、胶囊壳在溶出介质中的干扰,药物在溶出介质中的线性、回收率及稳定性等。在一些申报资料中,或者方法学验证内容不全面,或者虽进行了验证, 但所有试验不是在溶出介质中进行的,使审评人员难以判断溶出度方法的可行性。 6、 取样点和限度的确定:通过溶出度均一性试验( 考察同一批样品的溶出曲线)和重现性试验( 考察至少3批样品的溶出曲线),确定合理的 溶出度测定取样点和限度。为避免多次取样造成的误差,测定溶出曲线时取样点不宜过多,通常为5~6个点,小规格的制剂因采用100~250 ml溶出介质,所以溶出曲线一般可选3~4个时间点。限度应综合考虑溶出曲线拐点和一般性要求。 在新药审评的过程中发现,胶囊壳的干扰试验经常被申报单位忽视。在 USP26、BP2000和中国药典2005年版附录(公示稿)中,对空胶囊的干扰试验均有明确的规定,要求也基本一致。经试验考察,若空胶囊的干扰在2%以下时,可忽略不计;大于2%时,应对溶出度测定结果进行校正;大于25%时,则不能通过校正消除干扰, 溶出试验无效,应重新选择溶出度测定方法。 在溶出度研究资料中,另一个被忽视的问题是滤膜吸附情况的考察。在溶出度测定中,溶出液通常经过滤膜(中国药典规定滤膜孔径不大于0.8μm)滤过以得到澄清的溶液,试验所用滤膜应是惰性的,不能明显吸附溶出液中的有效成分,也不能被溶出介质溶出干扰测定的物质。因此在溶出度研究时,一定要首先考察试验所用滤膜对主成分是否有吸附,通常,吸附量在2%以下时可忽略不计; 超过2%时应考虑选用其他滤过方法或更换适宜的溶出介质。 另外在溶出度研究中还应注意溶出度试验仪的校正、溶出介质的脱气以及在操作中要严格执行SOP,保证实验数据的准确性,以全面正确和客观地反映药物的溶出情况。 以上是我个人对药品溶出度研究的几点认识 ,偏颇之处在所难免,欢迎大家批评指正。类别:审评二部转自:CDE 电子刊物

药物中残留溶剂甲醇含量的不确定度评定报告(*^__^*) 嘻嘻……,甲醇作为一种药物中常用的提取溶剂,已经在很多药物的提取中用到,譬如何首乌根甲醇提取,甘草的甲醇提取等都是天然药物的甲醇提取,还有一些生物药品的甲醇提取等等,已经屡见不鲜了的,俺一个同学课题中最近所用的一种有机药品分析,在溶剂的选择上就是该药品在甲醇中溶解性更好,在乙醇中溶解性差很多,也许因为甲醇极性比乙醇大的缘故吧,在日常的气相色谱和液相色谱分析中经常作为溶剂,不过甲醇又是一种有毒的醇类,因此,药物中的残留溶剂里面也就必须检测甲醇了的。俺们药物临床分析基地的老师前段时间就分析了一份某上级部门给的盲样,是分析该药品中的残留溶剂甲醇含量,报告结果的同时也要做甲醇的不确定度报告。俺只是个助手,(*^__^*) 嘻嘻……,下面就是此次残留溶剂甲醇的不确定度报告,请各位老师继续授业解惑。1材料与方法1.1仪器和试剂:日本岛津GC-15A气相色谱仪(FID检测器),色谱柱:GDX-102,2m*4mm,非极性色谱柱;柱温140℃,气化室200℃;检测室210℃;载气流50ml/min 。十万分之一电子天平,最大误差0.03mg;1.0ml移液管,允差±0.015ml;10ml容量瓶,允差±0.02ml;5ml容量瓶,允差±0.02ml;甲醇,纯度≥99.9%。1.2标准溶液:10ml容量瓶中加入约5ml蒸馏水,于十万分之一电子天平上称取甲醇10.10mg,稀释至刻度,制成1010ug/mL甲醇标准储备液。1.3考核样:国家某机构组织提供的考核盲样一支。1.4 检测方法:按照Ch.P.2010并参考药物临床分析基地醇类化合物检测的SOP要求进行操作。主要包括标准溶液配置,标准曲线制备,被测盲样解吸及样品解吸液上机测定,并根据标准曲线计算甲醇含量4个环节。2.测量结果的不确定度评定2.1 不确定度来源分析:主要采用国家药典分析方法测量甲醇的含量,故不考虑该方法本身的误差。在测量不确定度评定中只需考虑与实验操作有关的不确定度分量,主要包括以下4个方面:由标准曲线得解吸液中甲醇浓度、解吸液体积、标准溶液浓度对测量结果的影响、重复测量产生的不确定度即A类不确定度。2.1.1由标准曲线得解吸液中甲醇浓度的不确定度。用1010ug/ml甲醇标准储备液,配制出浓度分别为10.1、50.5、101、151.5、202、252.5ug/ml的6个标准溶液,其响应值测定结果见表1。采用最小二乘法拟合标准曲线时,计算得到解吸液中甲醇的浓度C的不确定度仅与峰面积的测量不确定度有关,标准溶液浓度不确定度的影响另行讨论。http://ng1.17img.cn/bbsfiles/images/2012/12/201212152020_412847_2355529_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/12/201212152020_412848_2355529_3.jpg由表1中的数据进行线性拟合得线性方程y=37.974x-90.421,γ=0.9998。测得解吸液中甲醇的浓度为158ug/mL。于是根据以上公式得标准不确定度u(x)=0.363ug/ml,相对标准不确定urel=0.0023。2.3 解吸液体积不确定度评定。解吸液体积不确定度分量包括刻度吸管校准不确定度和温度影响。实验用1.0ml移液管,以均匀分布估计,标准不确定度分量为0.009mL。温度的变化引起液体体积和量具容积的变化。但是由于液体的膨胀系数远远大于玻璃量具的膨胀系数。因此,可以忽略温度对量具容积的影响。当实验温度为23℃,以均匀分布估计,温度对体积测量的影响为0.001ml。经过汇总总解吸液体积的标准不确定度u(V)=0.0091ml,相对标准不确定度urel(V)=0.0091。2.4 标准溶液浓度不确定度。配制标准溶液先配制标准储备液,然后将标准储备液稀释成标准溶液。计算标准溶液浓度不确定度,应先计算标准储备液浓度不确定度,再计算标准溶液浓度不确定度。2.4.1标准储备液浓度不确定度来源,包括甲醇标准纯度影响、甲醇标准质量、溶液的体积。见表2http://ng1.17img.cn/bbsfiles/images/2012/12/201212152021_412851_2355529_3.jpg2.4.2配制标准溶液时量具校准引入的不确定度。见表3http://ng1.17img.cn/bbsfiles/images/2012/12/201212152022_412852_2355529_3.jpg2.4.3配制标准溶液时温差引入的不确定度。玻璃量器在20℃校准,配置溶液的温度为20±5℃,±5℃温度差引起的不确定度可通过估算温度范围和体积膨胀系数来进行计算。水的膨胀系数为2.1×10-4℃-1, 计算标准不确定度时假设温度变化是矩形分布,则在标准液逐级稀释过程中,由±5℃温差导致体积变化而引入的相对不确定度为:http://ng1.17img.cn/bbsfiles/images/2012/12/201212152022_412853_2355529_3.jpg2.5 A类不确定度评定 随机抽取相同条件下配制的甲醇含量相同的样品15个进行测定。按标准方法要求,样品经解吸后测定,由标准曲线计算解吸液中甲醇的浓度分别为158.75、160.04、156.09、158.44、155.70、160.07、158.01、162.46、159.78、159.01、158.33、155.72ug/mL,测定列的标准差为8.9ug/mL,本次试验测定样品数位一个,解吸液中甲醇浓度平均值为158ug/mL。则该测定结果的A类不确定度为: u[/size
[size=18px][b][b]1. 引言[/b] [/b]在药物制剂的研发及生产过程中,往往都会涉及到相关的药物粉体。这些粉体及其片剂的理化性质会影响其混合均匀度、压缩成型过程,以及最终制剂的生物利用度和疗效等,因此,在粉碎、混合、压片、制粒等过程中需要对其相关物理特性进行调控以确保最终制剂质量。除了关注度较高的粒度粒形,比表面积,流动性等性质外,密度及孔隙度的表征也是药物质量的重要指标,并且在研发及生产的众多环节都有所涉及。因而在美国药典USP 、USP ,日本药典JP 3.03,欧洲药典Ph. Eur. 2.9.32、Ph. Eur. 2.2.42和2020年版《中国药典》通用技术0992中,都明确规定了药物粉体相关的密度、孔隙度测定方法。密度主要会影响粉体的流动性,均匀性,压缩性以及离析度、结晶度等等。由片料包裹密度除以骨架密度算得的片料固相分数(Solid Fraction)是辊压过程中的关键工艺参数,测定固相分数可了解药物中固体含量百分比等相关信息,从而提高辊压过程的有效性,并建立可控的辊压速度、辊压压力等工艺操作参数,对工艺过程的参数设置及优化制剂质量具有重要意义。此外,药物材料的骨架密度还可以作为其结晶状态以及二元混合物比例的标志。孔隙度(Porosity)会影响药物的辊压制粒、崩解等过程,以及片剂强度、压实度、含量均匀度及溶出度等性质,是药物崩解、溶出和生物利用度的一个关键质量属性。此外,孔隙度测量还可以预测评估压缩过程中颗粒的变形特性,测量辊压后片料的总孔体积和固相分数,以及评估药物包衣的完整性,帮助确定包衣过程中物料流的参数设置等。综上所述,掌握和控制药物制剂的密度及孔隙度对药物的最终疗效及生产稳定性非常重要。本文将介绍药物粉体密度及孔隙度的定义及测试原理,并举例说明相关测试结果。[b][b]2. 密度测试[/b][/b]密度是单位体积粉体的质量。由于粉体的颗粒内部和颗粒间会存在空隙,所以粉体所占有的体积会因测量方法不同而有所差异,并由此产生如骨架密度、包裹密度等不同的密度概念。(1)真密度和骨架密度(颗粒密度)真密度也称绝对密度,所对应的真体积是指不包含开孔和闭孔的体积。骨架密度(颗粒密度)对应的骨架体积是样品的真实体积与闭孔体积之和,即不包括与外界连通的开孔体积。骨架密度的测定方法一般采用基于阿基米德原理的气体置换法测定,该法是目前世界公认的测真密度、骨架密度可靠的技术之一,并为无损测量。图1所示为麦克仪器的AccuPyc II[b]全自动气体置换法真密度仪[/b],测试采用惰性气体如氦气或氮气作为置换介质取代材料的孔隙体积,根据理想气体定律PV=nRT确定样品体积,结合样品质量可算得骨架密度。[/size][align=center][size=18px][img]http://img72.chem17.com/9/20200731/637318055225383925887.png[/img][/size][/align][size=18px][/size][align=center][size=18px]图1 AccuPyc II[/size][/align][size=18px][b]全自动气体置换法真密度仪[/b](2)包裹密度包裹密度所对应的包裹体积包含颗粒的骨架体积和开孔、闭孔体积,以及颗粒外表面的一些粗糙空隙。图2所示为麦克仪器的GeoPyc 1365[b]全自动包裹密度分析仪[/b]。包裹密度的测试原理是使用一种独特的替代测试技术,通常采用一种具备高流动性的微小刚性球状准流体介质作为替代介质将样品包裹起来。这种替代介质的颗粒很小,在混合过程中可与样品表面紧密贴合,但不会进入样品的孔隙中。[/size][align=center][size=18px][img]http://img75.chem17.com/9/20200731/637318055440362564765.png[/img][/size][/align][size=18px][/size][align=center][size=18px]图2 GeoPyc 1365[/size][/align][size=18px][b]全自动包裹密度分析仪[b]3. 孔隙度测试[/b] [/b]孔隙度指的是颗粒内的孔隙以及样品间隙所占体积与粉体体积之比,通常可通过压汞法和密度计算法等获得。孔隙度越高则表明药物中的总孔体积越大,对应的固体分数就越低。(1)压汞法压汞法是测量药物孔隙度特性常用的方法,可测得样品中与外界连通的开孔体积占总体积的百分比。压汞法的原理是基于汞对大多数固体材料不润湿,界面张力会抵抗汞进入孔中,要使得汞进入材料的开孔中则需要施加外部压力。汞压入的孔半径与所受外压成反比,根据Washburn方程可算出汞压入的孔半径与所受外力的对应关系。图3所示为麦克仪器的AutoPore V全自动压汞仪,其分析技术就是在[color=red]精确[/color]控制的压力下将汞压入材料的多孔结构中,通过测量不同外压下进入孔隙中汞的量,就可知道相应孔体积的大小。压汞法具有快速、高分辨率及分析范围广等优点,除了可测得孔隙度外,该表征还可获得样品的众多特性,例如:孔径分布、总孔体积、总孔比表面积、中值孔径等等。[/size][align=center][size=18px][img]http://img73.chem17.com/9/20200731/637318055737357739692.png[/img][/size][/align][align=center][size=18px]图3 AutoPore V[/size][/align][align=center][b][size=18px]全自动压汞仪[/size][/b][/align][size=18px](2)密度计算法除了压汞法外,通过将气体置换法真密度仪与包裹密度分析仪联用,结合材料的骨架密度和包裹密度,由式①也可直接计算出孔隙度。同时,由式②还可以算出片料的固体分数。[/size][align=center][size=18px][img]http://img74.chem17.com/9/20200731/637318055914530037790.jpg[/img][/size][/align][size=18px][/size][align=center][size=18px][img]http://img74.chem17.com/9/20200731/637318056110665447694.png[/img][/size][/align][size=18px]图4 AccuPyc II[b]全自动气体置换法真密度仪[/b]及GeoPyc 1365[b]全自动包裹密度分析仪[b]4. 密度及孔隙度测试举例[/b] [/b](1)药物辅料硬脂酸镁的骨架密度测定硬脂酸镁是新型药用辅料,可作固体制剂的成膜包衣材料、胶体液体制剂的增稠剂、混悬剂等。使用麦克仪器的AccuPyc II全自动气体置换法真密度仪对其进行骨架密度测试,结果表明,仪器在约16分钟内完成了10个测试循环,该硬脂酸镁样品的密度平均值为1.5157 g/cm3,标准偏差仅为0.0006 g/cm3,密度结果均围绕其平均值波动,结果非常稳定,实现了药物材料快速、高精度的体积测量和密度计算。(2)药物的压汞法孔隙度测定使用麦克仪器公司的AutoPore V [b]全自动压汞仪[/b]对某药物进行压汞测试。其堆积密度为1.1639 g/ml,骨架密度为1.5382 g/ml,由此计算得到的孔隙度为24.3332%。(3)药物片料的密度计算法孔隙度及固相分数测定使用麦克仪器的GeoPyc 1365[b]全自动包裹密度分析仪[/b]对辊压后得到的某药物片料进行孔隙度测试。测得该药物的包裹密度为1.3409 g/cm3,其标准偏差为0.0007 g/cm3,结合由AccuPyc II全自动气体置换法真密度仪测得的骨架密度1.4630 g/cm3,最后算得孔隙率为8.35 %。根据上文公式②,由骨架密度除以包裹密度可算得其固相分数为91.65 %。[b][b]5. 总结[/b][/b][/size][size=18px]药物粉体及相关制剂的密度及孔隙度表征对其处方设计、制备、质量控制等都具有重要指导意义。密度和孔隙度不仅是辊压和压片等过程的关键工艺参数,也是硬度、崩解度、溶出度、生物利用度等的关键质量属性,会直接影响和制约药物的性质及疗效。因而研究和掌握药物粉体及制剂的密度、孔隙度对获得高质量的药物至关重要。采用气体置换法真密度仪和包裹密度分析仪可分别获得药物粉体的骨架密度和包裹密度,通过压汞法或者结合两种密度仪的密度计算法可测得药物的孔隙度及片料的固体分数。借助这些性质表征有助于掌握及预测原料药及辅料在配方中的特性,评估药物制剂的批次变化及药物相关性能,从而优化制造过程和提升产品质量。[/size][size=18px][/size][size=18px][font=arial, helvetica, sans-serif][size=16px]关于麦克仪器公司[/size][/font][font=arial, helvetica, sans-serif][size=16px]麦克仪器公司是专业提供表征颗粒,粉体和多孔材料的物理性能,化学活性和流动性的高性能设备的全球领先的生产商。我们的技术包括:比重密度法、吸附、动态化学吸附、颗粒大小和形状、压汞孔隙度测定、粉末流变学和催化剂活性测试。公司在美国、英国和西班牙设有研发和生产基地,并在美洲、欧洲和亚洲设有直销和服务业务。麦克仪器是创新性的公司,产品是著名的政府和学术机构的10,000多个实验室的首选仪器。我们拥有世界一流的科学家和积极响应的支持团队,通过将Micromeritics技术应用于客户的需求,帮助客户获得成功。更多信息,请访问: [/size][/font][url=http://www.micromeritics.com.cn/][color=#0000ff][font=arial, helvetica, sans-serif]www.micromeritics.com.cn [/font][/color][/url][/size]
[center]跨国药企上书 外资药企争议基本药物制度[/center]虽然新医改方案征求意见的截止日期已过,但来自各方的建议仍源源不断汇入相关部门。3日,记者获得中国外商投资企业协会药品研制和开发行业委员会(以下简称RDPAC)对于医改征求意见稿的一份建议书,RDPAC的执行总裁萧滋杰也接受了本报记者的专访,详述对于新医改方案的认同和不认同,特别对于基本药物制度的执行,提出一些不同的看法。 外资药企,是中国医药市场上的一股重要力量,占据了处方药市场30%的市场份额。这个群体对于新医改的影响力不容忽视。而RDPAC,正是跨国药企在华的代言人。争议基本药物社区全覆盖在新医改方案中,建立国家基本药物制度是一项重要内容。所谓基本药物,是国家按照“防治必需、安全有效、价格合理、使用方便、中西药并重”的原则,结合我国用药特点并参照国际经验,确定的药物通用名目录。事实上,基本药物目录在我国一直存在,但一直以来形同虚设,未起到实际作用。为此,新医改旨在通过建立一系列制度保障基本药物目录的运行。 征求意见稿对基本药物制度做出如下阐述:中央政府统一制定和发布国家基本药物目录。基本药物由国家实行招标定点生产或集中采购,直接配送,在合理确定生产环节利润水平的基础上统一制定零售价。这也正是被外界诟病的“统购统销”。——此前,代表民族药企的中国医药企业管理协会已经上书发改委等相关部门,对此似乎违背市场经济运行的做法表示了不同的意见。“在这一点上,我们也是同样的看法。”萧滋杰说,按照招标定点生产,有悖国家反垄断的原则,会让少数垄断企业获益,但危及大部分企业的生存。他认为,对基本药物的生产和流通企业不宜作单一的定点或招标限定,可以在政府的有效监管下鼓励企业合理竞争,通过竞争性议价或团购方式来遴选药品供销企业。 而基层医疗卫生机构全部使用基本药物的新政,更让跨国药企坐立不安。征求意见稿明确指出,城市社区卫生服务中心(站)、乡镇卫生院、村卫生室等基层医疗卫生机构,应全部使用基本药物,其他各类医疗机构也要将基本药物作为首选药物并确定使用比例。这让一直试图拓展基层医疗市场的跨国药企发现,自己生产的中高价药物可能无法拿到进入这一市场的敲门砖。值得一提的是,新医改下,社区医疗等基层医疗市场未来增长潜力巨大,这一背景下,众多跨国药企一改往日只重视三甲医院的营销,转而向社区拓展,但如果社区只准使用基本药物,大部分跨国药企可能都将和这一未来扩容最大的市场无缘。对此,萧滋杰表示,此种做法并不合理。对社区医院用药的限制与拓展社区医疗的初衷背道而驰。“国家拓展社区医疗,是为了从大医院分流病患。但如果患者和医生在社区医院没有了用药的选择权,很多患者可能会再次回流到三级医院。
一、概述药物中的残留溶剂系指在原料药或辅料的生产中、以及在制剂制备过程中使用或产生而又未能完全去除的有机溶剂。根据国际化学品安全性纲要,以及美国环境保护机构、世界卫生组织等公布的研究结果,很多有机溶剂对环境、人体都有一定的危害,因此,为保障药物的质量和用药安全,以及保护环境,需要对残留溶剂进行研究和控制。本指导原则是在参考人用药物注册技术要求国际协调会(International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use,ICH)颁布的残留溶剂研究指导原则,美国药典(the United States Pharmacopoeia,USP)、英国药典(British Pharmacopoeia, BP)、欧洲药典(European Pharmacopoeia,EP)、中国药典(Chinese Pharmacopoeia, ChP)相关内容的基础上,结合我国药物研发的特点,通过分析、研究残留溶剂问题与药物的安全性、有效性及质量可控性之间的内在关系而制定的。本指导原则总结了对残留溶剂问题的一般认识,旨在帮助药物研发者科学合理的进行残留溶剂方面的研究,也为药物评价者提供参考。考虑到残留溶剂研究涉及的范围比较广泛,本指导原则主要对原料药的残留溶剂问题进行讨论,并以此为基础,探讨和总结药物研究过程中对残留溶剂问题的一般性原则。药物研发者可参考本指导原则对制剂和辅料的残留溶剂问题进行研究。考虑到药物研究开发的阶段性,本指导原则适用于药物研发的整个过程。
我刚接到一个pH依赖性的药物,我想问问,溶解度曲线该怎么做呢?
发布日期20050323关于普通口服制剂溶出度比较研究的一些建议当药品处方、生产工艺、生产地点和生产规模等发生变更后,最需要验证的问题就是变更前后产品质量是否保持一致。对于口服固体制剂而言,溶出度或释放度对比研究是比较变更前后产品相似性或差异程度的一个重要工具。为保证该对比研究能提供有效的信息,首先此项研究需要结合药物的生物学性质及制剂特性展开,其次要采用合理的方法对研究结果进行统计分析。本文将针对普通口服制剂的溶出对比研究提出一些建议。一、 实验方法 为保证测定结果具有一定的统计意义,并且尽可能减少其他变量的影响,试验中需关注以下问题: (1)变更前后样品测试需采用相同的仪器,尽可能在同一天进行。 (2)一般每批样品至少采用12个剂量单位(如片剂为12片,胶囊为12粒)进行测定。除0时外,一般至少选择3个时间点进行测定,如5、15、30、45min,或采用其他适宜的时间间隔取样,直到药物溶出90%以上或达到溶出平台,计算各时间点药物溶出百分比,绘制每批样品药物溶出曲线。 (3)除0时外,第1个时间点溶出结果的变异系数不得过20%,从第2个时间点至最后1个时间点的溶出结果的变异系数应小于10%。 下面根据原料药生物学性质的不同,分类阐述: 1.原料药属于高溶解性,高渗透性的 此类药物溶出比较建议首先选择在900mL0.1N HCl中进行,可采用药典收载的转蓝法(转速100rpm),也可选择药典收载的桨法(转速50rpm)。如果15分钟内(一般认为餐后胃平均保留T50%是15-20分钟)药物溶出85%以上,则不需要再比较其他pH条件下或介质中药物溶出情况。如果15分钟内药物溶出未达到85%,则需要按下述2或3对变更前后溶出行为进行比较。 2.原料药属于高溶解性,低渗透性的 此类药物由于渗透性低而溶解性好,药物的渗透性是体内吸收的限速步骤,而主要不取决于制剂的溶出。因此, 一般不需要在不同PH条件下考察产品变更前后溶出情况。溶出比较研究可选择质量标准中规定的检查方法进行,如标准中未收载溶出度检查方法,可选择产品申请上市注册时质量研究和稳定性考察中选择的溶出度检查方法。 3.原料药属于低溶解性、高渗透性的 由于此类药物渗透性高,药物的溶出过程可能是体内吸收的限速步骤,因此,建议考察不同PH条件下变更前后产品溶出情况,可选择水、0.1N盐酸及pH 4.5-7.5缓冲液三种介质进行比较。对于胶囊或含明胶包衣的片剂,可采用含酶的人工胃液或人工肠液进行。如无特殊情况,溶出比较研究一般不使用含有机溶剂的介质(如乙醇-水体系)进行。如有充分的依据,介质中可使用适量的表面活性剂。如果原料药或处方中辅料属于PH非敏感型的,溶出曲线比较可仅采用2种缓冲体系进行。 二、统计分析方法 变更前后溶出曲线比较可采用适宜的统计学方法进行。 溶出曲线比较可采用模型依赖法,即一些用于描述药物溶出曲线的数学模型,如线性模型、Weibull模型等。进行此类统计学比较,一般先根据变更前的代表性批次的溶出曲线,选择最合适的模型,建议采用不超过三个参数的模型(例如线性、二次方、对数、概率和Weibull模型),再对模型的参数进行统计学比较,基于对已批准的标准批次的测试单位(例如胶囊或片)的匹配模型的参数变化设置相似区间,以测试批次和参比批次间的模型参数计算MSD,估计在两个批次间真实差别的90%可信区间,比较可信区间和相似区间的限度。如果可信区间是在相似区间的范围内,则认为测试批次与参比批次有相似的溶出曲线。 溶出曲线比较也可选择非模型依赖方法,如可通过计算相似因子f2比较变更前后溶出行为的相似性,当f2数值在50-100范围认为两条溶出曲线是相似的。 f2=50log 上述公式中n为时间点(n≥3),Rt是变更前制剂药物溶出平均百分数,Tt是变更后制剂药物溶出平均百分数。采用相似因子比较法需满足以下条件: ● 取样时间点除0时外,至少有3个 ● 每个处方样品至少采用12个剂量单位 ● 只能有一个时间点药物溶出达到85%以上 ● 从第2个时间点至最后1个时间点溶出结果的变异系数应小于10% ●保证药物溶出90%以上或达到溶出平台 如果药物在15min内溶出达到85%以上,可以认为两批产品溶出行为是相似的,不需要通过统计学方法对数据分析判定。 【附注】 1 药物的水溶解性 主要反应药物在生理PH条件下的溶解性情况。研究工作一般在37±1℃条件下,pH1—7.5的水性介质中进行测定,绘制被测药物的pH—溶解度曲线。可根据药物的离子化特性选择pH测定点,例如,当药物的pka为3—5时,药物的溶解度建议在pH=pka, pH=pka+1, pH=pka-1, pH=1和pH=7.5处测定。药典收载的缓冲溶液可用于药物溶解度的研究。 药物水溶解性可根据pH1—7.5范围溶解药物单次最大给药剂量的介质的体积来决定。在pH1—7.5范围,如果单次最大给药剂量的药物可溶于不多于250 ml的介质中,则该药物认为是高溶解性的。 2 药物的渗透性 药物渗透性分类以测定药物透过人体肠壁膜量为直接依据,而药物在人体吸收程度(指药物吸收比例,而不是系统生物利用度)只是间接依据。 药物渗透性的测定可采用人体实验方法或其他能预测药物体内吸收程度的非人体实验方法。人体实验方法包括质量平衡法、绝对生物利用度法和小肠灌流法等。利用未标记稳定同位素或标记放射性同位素进行的药物药代动力学质量平衡研究可以反映药物的吸收程度,但对多数药物研究结果显示,此方法测定结果变异程度大,一般优先考虑采用其它方法。以静脉注射给药为对照,测定口服给药的绝对生物利用度某些情况下可以间接反映药物吸收情况,如无法证明药物在胃肠道内是否稳定的情况下,药物吸收程度达到90%以上时,该药物被认为是高渗透性的。其他能预测药物体内吸收程度的非人体实验方法也可作为判定药物渗透性的依据,如使用适宜的动物模型进行体内或在体灌肠研究,使用人或动物肠组织切样进行体外渗透性实验,体外表皮单细胞培养通透性实验。上述实验可参照相关技术指导原则进行。考虑药物在透过胃肠壁膜前可能会有部分降解,为证明药物从胃肠道消除是由于药物透过胃肠壁膜而不是发生降解,研究中需注意考察药物在胃肠道中的稳定性。多数情况下,一种实验方法已经可以说明药物的渗透性(如90%以上的药物可在尿中回收)。当一种方法不能确定药物的通透性时,可用两种不同的方法。此外,药物的化学结构或某种理化性质(如分配系数等)也可为药物渗透性提供有用的信息。 对于前体药物,其渗透性取决于前体药物向药物转化的机制和部位。如果前药在透过肠壁膜后转化为药物,则需测定前药的渗透性;反之,如果前药在胃肠道内已就转化为药物,则应测定药物的渗透性【参考文献】1、Dissolution Testing of Immediate Release Solid Oral Dosage Forms. U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER).
[b]第一法 仪器装置[/b] (1)转篮分篮体与篮轴两部分,均为不锈钢金属材料制成。篮体A由不锈钢丝网(丝径为0.254mm,孔径0.425mm)焊接而成,呈圆柱形,内径为22.2±1.0mm,上下两端都有金属边缘。篮轴B的直径为9.4~10.1mm,轴的末端连一金属片,作为转篮的盖;盖上有通气孔(孔径2.0mm);盖边系两层,上层外径与转篮外径同,下层直径与转篮内径同 盖上的三个弹簧片与中心呈120°角。转篮旋转时摆动幅度不得超过±1.0mm。 (2)操作容器为1000ml的圆底烧杯,内径为98~106mm,高160~175mm;烧杯上有一有机玻璃盖,盖上有2孔,中心孔为篮轴的位置,另一孔供取样或测温度用。为使操作容器保持恒温,应外套水浴;水浴的温度应能使容器内溶剂的温度保持在37±0.5℃。转篮底部离烧杯底部的距离为25±2mm。 (3)电动机与篮轴相连,转速可任意调节在每分钟50~200转,稳速误差不超过±4%。运转时整套装置应保持平稳,不得晃动或振动。 (4)仪器应装有6套操作装置,可一次测定6份供试品。取样点位置应在转篮上端距液面中间,离烧杯壁10mm处。 测定法:除另有规定外,量取经脱气处理的溶剂900ml,注入每个操作容器内,加温使溶剂温度保持在37±0.5℃,调整转速使其稳定。取供试品6片(个),分别投入6个转篮内,将转篮降入容器中,立即开始计时,除另有规定外,至45分钟时,在规定取样点吸取溶液适量,立即经0.8μm微孔滤膜滤过,自取样至滤过应在30秒钟内完成。取滤液,照各药品项下规定的方法测定,算出每片(个)的溶出量。 结果判断 6片(个)中每片(个)的溶出量,按标示含量计算,均应不低于规定限度(Q);除另有规定外,限度(Q)为标示含量的70%。如6片(个)中仅有1~2片(个)低于规定限度,但不低于Q-10%,且其平均溶出量不低于规定限度时,仍可判为符合规定。如6片(个)中有1片(个)低于Q-10%,应另取6片(个)复试;初、复试的12片(个)中仅有1~2片(个)低于Q-10%,且其平均溶出量不低于规定限度时,亦可判为符合规定。供试品的取用量如为2片(个)或2片(个)以上时,算出每片(个)的溶出量,均不得低于规定限度(Q);不再复试。[b]第二法 仪器装置 [/b] 除将转篮换成搅拌桨(A)外,其他装置和要求与第一法同。 搅拌桨由不锈钢金属材料制成。旋转时摆动幅度A、B不得超过±0.5mm。取样点应在桨叶上端距液面中间,离烧杯壁10mm处。 测定法:除另有规定外,量取经脱气处理的溶剂900ml,注入每个操作容器内,加温使溶剂温度保持在37±0.5℃。取供试品6片(个),分别投入6个操作容器内(用于胶囊剂测定时,如胶囊上浮,可用一小段耐腐蚀的金属线轻绕于胶囊外壳),立即启动旋转并开始计时,除另有规定外, 至45分钟时,在规定取样点吸取溶液适量,立即经0.8μm微孔滤膜滤过,自取样至滤过应在30秒钟内完成。取滤液,照各药品项下规定的方法测定,算出每片(个)的溶出量。 结果判断 同第一法。[b]第三法 仪器装置 [/b] (1)搅拌桨 由不锈钢制成;桨杆上部直径为9.4~10.1mm,桨杆下部直径为6.0±0.2mm,旋转时摆动幅度A、B不得超过±0.5mm,取样点应在桨叶上端距液面中间,离烧杯壁6mm处。桨叶底部离烧杯底部的距离为15±1mm。 (2)操作容器为250ml的圆底烧杯,内径为62±3mm,高为126±6mm,烧杯上有一有机玻璃盖,盖上有一开口,为放置搅拌桨、取样及测温用。其他要求同第一法(2)。 (3)电动机与桨杆相连,转速可任意调节在每分钟25~100转,稳速误差不超过±1转。 动转时整套装置应保持平稳,不得晃动或振动。 测定法 除另有规定外,量取经脱气处理的溶剂100~250ml注入每个操作容器内,以下操作同第二法。 结果判断 同第一法。







 我要推广仪器
我要推广仪器
 下载APP
下载APP