求实验室采购的用于微量、痕量物质色谱分析所用的有机试剂的空白试验操作作业指导书
求实验室采购的用于微量、痕量物质色谱分析所用的有机试剂的空白试验操作作业指导书
国标中空白试验的定义空白试验:除不加试样外,采用完全相同的分析步骤、试剂和用量(滴定法中标准滴定液的用量除外),进行平行操作所得的结果。用于扣除试样中试剂本底和计算方法的检出限。空白做不好会怎样?空白试验测得的结果称为空白试验值。在进行样品分析时所得的值减去空白试验值得到的才是最终分析结果。空白试验是实验室质量控制的重要环节,其准确性对提高检测结果的准确度至关重要。空白试验值反应了测试仪器的噪声、试剂中的杂质、环境及操作过程中的沾污等因素对样品测定产生的综合影响,直接关系到测定的最终结果的准确性。空白试验值低,数据离散程度小,分析结果的精度随之提高,它表明分析方法和分析操作者的测试水平较高。当空白试验值偏高时,应全面检查试验用水、试剂、量器和容器的沾污情况、测量仪器的性能及试验环境的状态等,以便尽可能地降低空白试验值。做酸碱滴定时为什么要做空白试验?因为用作稀释液的空白溶液有一定的酸碱度,会影响滴定结果,所以要做空白试验,来扣除空白的干扰。比如空白溶液为酸性,这时候用碱滴定此溶液,得到的结果会偏高,要扣除空白溶液的酸度,得到的酸度才正确。[b][color=#ac1d10]石墨炉测铅时,空白[color=#888888](4硝酸+1高氯酸GR)[/color]值总是较高,与灰化法的结果不大一致(样品为植物样)。[/color][/b][color=#595959]【分析】1. 所用的水为用石英亚沸水蒸馏器蒸馏得到的,先打一下空白,一般不会超过0.0015,然后采用的为硝酸(工艺超纯)和高氯酸(优极纯)消化,最后溶解用的1摩尔每升的盐酸或硝酸(结果差不多,只是盐酸稳定性要好一点),定容体积为50mL的话空白值一般为0.03左右。不过铅比较难做,基体干扰很大。[/color][color=#595959]2. 空白问题来自多方面,上面说的水与试剂外,你用的氩气是高纯的吗?也可用高纯氮气,但要注意分子带背景[/color][color=#595959]3. 可能来自由你所用的硝酸和高氯酸不纯所致。你可以先测空管,然后测你所用的水,再测含酸的水空白看看就知道了[/color][color=#595959]4.有可能是试液的酸度过大会影响测定,特别是使用高氯酸,影响更大,酸度大对石墨炉损害也比较大。对于石墨炉测定铅,湿法消化最好使用微波消化,使用硝酸和双氧水,这样空白中酸度比较容易控制,空白也比较低。[/color][color=#595959]5. a.实际Pb含量有出入,厂家就没有测准;b.你的仪器可能没有调制最佳。[/color]原子荧光空白偏高原因1.测定介质的选择及浓度的影响选择HNO3、HCl、HClO4、H3PO4和H2SO4等进行了实验,结果表明:以HClO4和H3PO4作介质响应值较低,汞在H2SO4溶液中能得到较大灵敏度,但空白值也高。汞在HNO3和HCl溶液中的荧光强度差别不大。在5%一25%的硝酸和5%一20%的盐酸中荧光强度基本稳定。[b][/b]2.酸的影响作为原子荧光的载流和标准试剂的基体,酸对原子荧光的影响十分的突出。一般在原子荧光中使用的酸主要有两种-盐酸和硝酸。我们习惯上使用盐酸。如果购买市场上质量差的酸,对空白的影响要大得多。[b][/b]3.还原剂的影响作为原子荧光的还原剂,一般使用最多的是硼氢化钾或硼氢化钠,在原子荧光法中,还原剂对样品以及空白值的影响主要体现在它的用量。硼氢化钾浓度不宜超过3.0%。实验表明,当硼氢化钠浓度为1%时,灵敏度和稳定性好,并且有效消除干扰。[b][/b]4.灯电流的影响一般来说灯电流与负高压对原子荧光的影响是同方向的。提高灯电流,空白值就相应的提高。空白值太大的话,可以适当的降低电流值,使仪器的灵敏度降低,减少标准空白的背景值,使所测的数据准确、可靠,但是如果电流过小,灵敏度也将变小,对所测样品的影响就会增加,所以选择合适的灯电流,保证一定的空白值与灵敏度才是关键。[b][/b]5.载气的影响实验表明,当氩气流速较低时,测定的灵敏较高.但结果的变动性加大,实际测定取氩气流速200ml/hifn,此时测定的灵敏度较高,相对标准偏差可以接受。空白色谱柱出峰,什么原因?[align=center][/align][i][color=#595959]高效液相色谱,色谱柱是Kromasil C18(15cm*5Um),流动相为10%乙腈和90%三氟乙酸水,走空白色谱柱时在整个保留时间的中间位置出现较大的色谱峰,走了好几针空白,每针的响应都不同,有的很高,有的又很低,但杂峰一直存在,不像是色谱柱里有杂质,用流动相冲洗后依然出现此问题,什么原因?[/color][/i][b][color=#595959][/color][color=#3e3e3e]【原因】[/color][/b][color=#3e3e3e]1、确定检测波长,如果是末端吸收,排除三氟乙酸的干扰。[/color][color=#3e3e3e]2、使用100%乙腈作为流动相,乙腈作为样品进样,如果为基线,则说明色谱柱及系统完好。[/color][color=#3e3e3e]3、然后在换流动相为10%乙腈和90%三氟乙酸水,样品为乙腈,如果有上述“中间位置出现较大的色谱峰”,那说明八成问题在三氟乙酸上,或者稀释三氟乙酸的水上。[/color][color=#3e3e3e]4、如果上述2、3都有“中间位置出现较大的色谱峰”,检查检测器及进样器。[/color][color=#3e3e3e]5、这些还不能解决,请联系工程师[/color][color=#3e3e3e][/color]空白乙腈出峰,什么原因?[i][color=#595959]岛津高效液相色谱仪,进空白乙腈,在样品峰位子出现倒峰,进双蒸水在样品峰出现正峰,进样品时有杂峰干扰,我已经排除了水和乙腈的问题,不知道是不是进样池或者检测器污染了,已经整了10多天,还是没效果,请指教原因和解决办法。[/color][/i][b]【分析】[/b]液相中出现倒峰,主要原因是样品吸收比流动相吸收小,所以排除影响,不仅要考虑进样系统污染和检测池污染,还要考虑流动相和色谱柱的影响。换一根柱子试试,考察系统的情况,如果照旧,就排除色谱柱的影响,然后更换流动相试试,再以后清洗进样系统和检测池等等。或将色谱柱从系统中断开,直接用乙腈或水做流动相清洗检测器就可以知道是否是检测器受污染,如果基线正常,再检查你的进样系统,样品,前处理是否有问题,如果都没问题,就去检查色谱柱吧。[color=#ac1d10][b]水质分析中氨氮的测定空白值很高的原因[/b][/color][align=center][/align]实验中纳氏试剂和酒石酸钾钠对空白值的高低影响很大,可能原因如下:1.测定使用的蒸馏水被污染,含有影响实验结果的杂质,比如蒸馏水含氨。2.试管、移液管等仪器没有清洗彻底,这些因素对实验结果也有较大的影响。3.实验所用的无氨水质量不佳,含氨量比较高,导致实验得到的空白值偏高,这是最主要的原因。4.在配制酒石酸钾钠和钠氏试剂时出现较大偏差,导致实验所用的试剂纯度(酒石酸钾钠、钠氏试剂)不佳,从而导致实验得到的空白值偏高。总氮空白值偏高,什么原因?1、试剂的配制碱性过硫酸钾的配制过程十分重要,掌握不好,会影响消解效果,对测定结果产生一定的影响。配制该溶液时,可分别称取过硫酸钾和氢氧化钠,两者分开配制,再混合定容,或者先配制氢氧化钠溶液,待其温度降到室温后再加入过硫酸钾溶解。若二者在一只烧杯中溶于水,应缓慢加水,同时搅拌,防止氢氧化钠放热使溶液温度过高引起局部过硫酸钾失效。2、玻璃器皿的洗涤[b][/b]所使用的玻璃器皿应先用(1+9)盐酸浸泡后,再用无氨水冲洗数次才能使用,否则,也会造成空白值偏高或平行性较差的情况。3、比色时的注意事项该项目的测定涉及两个波长(220nm和275nm),建议在测定完一组样品的同一波长后,再调整到另一波长,统一测定,不要测完一个样品的两个吸光度后再换另一个样品,这样反复调整波长会引起一定的测量误差。4、试剂的选择碱性过硫酸钾消解紫外分光光度法测定总氮的过程中,过硫酸钾是至关重要的试剂。首先,试剂的纯度关系到空白值的高低、测定结果的准确度。一般普通分析纯过硫酸钾的总氮含量最高不超过0.005%,但由于试剂质量存在差异,有些厂家、批次的试剂含氮量常常达不到这个要求,致使空白值偏高。因此,有条件的话建议使用优级纯试剂,尽量降低试剂中的含氮量,从而降低实验空白值。5、实验用水及试剂的质量检验[b][/b]若实验的空白值不够理想,则需要对实验用水及试剂进行检验,以选择出含氮量最低的水和试剂,获得理想的空白值。[color=#ac1d10][b]粗蛋白测定空白偏高的影响因素?[/b][/color]可以从以下方面考虑:1.配溶液的水换没换;2.看看消化炉的回收率;3.看看蒸馏仪的回收率;4.盐酸在不在保质期内;5.配溶液的硫酸是不是有问题;6.检查催化剂,是不是含有硝酸钾。小结空白试验是实验室质量控制的重要环节,其准确性对提高检测结果的准确度至关重要。以分光光度法空白为例 ,要用相应的实验用纯水或有机溶济作参比,不能用空白实验溶液做参比,以免人为减小空白值,从而掩盖空白实验值的变异 。值得注意的是,通过计量认证的实验室,由于检测仪器和玻璃量器定期检定 ,以及采取了全面的质量管理措施,因此,若测定的空白实验值太大,一般是化学药品纯度不够,配制的试液变质或被污染等原因 。应重新配制试液 ,返工重测该批样品 ,直至空白实验值合格。国标中空白试验的定义空白试验:除不加试样外,采用完全相同的分析步骤、试剂和用量(滴定法中标准滴定液的用量除外),进行平行操作所得的结果。用于扣除试样中试剂本底和计算方法的检出限。空白做不好会怎样?空白试验测得的结果称为空白试验值。在进行样品分析时所得的值减去空白试验值得到的才是最终分析结果。空白试验是实验室质量控制的重要环节,其准确性对提高检测结果的准确度至关重要。空白试验值反应了测试仪器的噪声、试剂中的杂质、环境及操作过程中的沾污等因素对样品测定产生的综合影响,直接关系到测定的最终结果的准确性。空白试验值低,数据离散程度小,分析结果的精度随之提高,它表明分析方法和分析操作者的测试水平较高。当空白试验值偏高时,应全面检查试验用水、试剂、量器和容器的沾污情况、测量仪器的性能及试验环境的状态等,以便尽可能地降低空白试验值。做酸碱滴定时为什么要做空白试验?因为用作稀释液的空白溶液有一定的酸碱度,会影响滴定结果,所以要做空白试验,来扣除空白的干扰。比如空白溶液为酸性,这时候用碱滴定此溶液,得到的结果会偏高,要扣除空白溶液的酸度,得到的酸度才正确。[b][color=#ac1d10]石墨炉测铅时,空白[color=#888888](4硝酸+1高氯酸GR)[/color]值总是较高,与灰化法的结果不大一致(样品为植物样)。[/color][/b][color=#595959]【分析】1. 所用的水为用石英亚沸水蒸馏器蒸馏得到的,先打一下空白,一般不会超过0.0015,然后采用的为硝酸(工艺超纯)和高氯酸(优极纯)消化,最后溶解用的1摩尔每升的盐酸或硝酸(结果差不多,只是盐酸稳定性要好一点),定容体积为50mL的话空白值一般为0.03左右。不过铅比较难做,基体干扰很大。[/color][color=#595959]2. 空白问题来自多方面,上面说的水与试剂外,你用的氩气是高纯的吗?也可用高纯氮气,但要注意分子带背景[/color][color=#595959]3. 可能来自由你所用的硝酸和高氯酸不纯所致。你可以先测空管,然后测你所用的水,再测含酸的水空白看看就知道了[/color][color=#595959]4.有可能是试液的酸度过大会影响测定,特别是使用高氯酸,影响更大,酸度大对石墨炉损害也比较大。对于石墨炉测定铅,湿法消化最好使用微波消化,使用硝酸和双氧水,这样空白中酸度比较容易控制,空白也比较低。[/color][color=#595959]5. a.实际Pb含量有出入,厂家就没有测准;b.你的仪器可能没有调制最佳。[/color]原子荧光空白偏高原因1.测定介质的选择及浓度的影响选择HNO3、HCl、HClO4、H3PO4和H2SO4等进行了实验,结果表明:以HClO4和H3PO4作介质响应值较低,汞在H2SO4溶液中能得到较大灵敏度,但空白值也高。汞在HNO3和HCl溶液中的荧光强度差别不大。在5%一25%的硝酸和5%一20%的盐酸中荧光强度基本稳定。[b][/b]2.酸的影响作为原子荧光的载流和标准试剂的基体,酸对原子荧光的影响十分的突出。一般在原子荧光中使用的酸主要有两种-盐酸和硝酸。我们习惯上使用盐酸。如果购买市场上质量差的酸,对空白的影响要大得多。[b][/b]3.还原剂的影响作为原子荧光的还原剂,一般使用最多的是硼氢化钾或硼氢化钠,在原子荧光法中,还原剂对样品以及空白值的影响主要体现在它的用量。硼氢化钾浓度不宜超过3.0%。实验表明,当硼氢化钠浓度为1%时,灵敏度和稳定性好,并且有效消除干扰。[b][/b]4.灯电流的影响一般来说灯电流与负高压对原子荧光的影响是同方向的。提高灯电流,空白值就相应的提高。空白值太大的话,可以适当的降低电流值,使仪器的灵敏度降低,减少标准空白的背景值,使所测的数据准确、可靠,但是如果电流过小,灵敏度也将变小,对所测样品的影响就会增加,所以选择合适的灯电流,保证一定的空白值与灵敏度才是关键。[b][/b]5.载气的影响实验表明,当氩气流速较低时,测定的灵敏较高.但结果的变动性加大,实际测定取氩气流速200ml/hifn,此时测定的灵敏度较高,相对标准偏差可以接受。空白色谱柱出峰,什么原因?[align=center][/align][i][color=#595959]高效液相色谱,色谱柱是Kromasil C18(15cm*5Um),流动相为10%乙腈和90%三氟乙酸水,走空白色谱柱时在整个保留时间的中间位置出现较大的色谱峰,走了好几针空白,每针的响应都不同,有的很高,有的又很低,但杂峰一直存在,不像是色谱柱里有杂质,用流动相冲洗后依然出现此问题,什么原因?[/color][/i][b][color=#595959][/color][color=#3e3e3e]【原因】[/color][/b][color=#3e3e3e]1、确定检测波长,如果是末端吸收,排除三氟乙酸的干扰。[/color][color=#3e3e3e]2、使用100%乙腈作为流动相,乙腈作为样品进样,如果为基线,则说明色谱柱及系统完好。[/color][color=#3e3e3e]3、然后在换流动相为10%乙腈和90%三氟乙酸水,样品为乙腈,如果有上述“中间位置出现较大的色谱峰”,那说明八成问题在三氟乙酸上,或者稀释三氟乙酸的水上。[/color][color=#3e3e3e]4、如果上述2、3都有“中间位置出现较大的色谱峰”,检查检测器及进样器。[/color][color=#3e3e3e]5、这些还不能解决,请联系工程师[/color][color=#3e3e3e][/color]空白乙腈出峰,什么原因?[i][color=#595959]岛津高效液相色谱仪,进空白乙腈,在样品峰位子出现倒峰,进双蒸水在样品峰出现正峰,进样品时有杂峰干扰,我已经排除了水和乙腈的问题,不知道是不是进样池或者检测器污染了,已经整了10多天,还是没效果,请指教原因和解决办法。[/color][/i][b]【分析】[/b]液相中出现倒峰,主要原因是样品吸收比流动相吸收小,所以排除影响,不仅要考虑进样系统污染和检测池污染,还要考虑流动相和色谱柱的影响。换一根柱子试试,考察系统的情况,如果照旧,就排除色谱柱的影响,然后更换流动相试试,再以后清洗进样系统和检测池等等。或将色谱柱从系统中断开,直接用乙腈或水做流动相清洗检测器就可以知道是否是检测器受污染,如果基线正常,再检查你的进样系统,样品,前处理是否有问题,如果都没问题,就去检查色谱柱吧。[color=#ac1d10][b]水质分析中氨氮的测定空白值很高的原因[/b][/color][align=center][/align]实验中纳氏试剂和酒石酸钾钠对空白值的高低影响很大,可能原因如下:1.测定使用的蒸馏水被污染,含有影响实验结果的杂质,比如蒸馏水含氨。2.试管、移液管等仪器没有清洗彻底,这些因素对实验结果也有较大的影响。3.实验所用的无氨水质量不佳,含氨量比较高,导致实验得到的空白值偏高,这是最主要的原因。4.在配制酒石酸钾钠和钠氏试剂时出现较大偏差,导致实验所用的试剂纯度(酒石酸钾钠、钠氏试剂)不佳,从而导致实验得到的空白值偏高。总氮空白值偏高,什么原因?1、试剂的配制碱性过硫酸钾的配制过程十分重要,掌握不好,会影响消解效果,对测定结果产生一定的影响。配制该溶液时,可分别称取过硫酸钾和氢氧化钠,两者分开配制,再混合定容,或者先配制氢氧化钠溶液,待其温度降到室温后再加入过硫酸钾溶解。若二者在一只烧杯中溶于水,应缓慢加水,同时搅拌,防止氢氧化钠放热使溶液温度过高引起局部过硫酸钾失效。2、玻璃器皿的洗涤[b][/b]所使用的玻璃器皿应先用(1+9)盐酸浸泡后,再用无氨水冲洗数次才能使用,否则,也会造成空白值偏高或平行性较差的情况。3、比色时的注意事项该项目的测定涉及两个波长(220nm和275nm),建议在测定完一组样品的同一波长后,再调整到另一波长,统一测定,不要测完一个样品的两个吸光度后再换另一个样品,这样反复调整波长会引起一定的测量误差。4、试剂的选择碱性过硫酸钾消解紫外分光光度法测定总氮的过程中,过硫酸钾是至关重要的试剂。首先,试剂的纯度关系到空白值的高低、测定结果的准确度。一般普通分析纯过硫酸钾的总氮含量最高不超过0.005%,但由于试剂质量存在差异,有些厂家、批次的试剂含氮量常常达不到这个要求,致使空白值偏高。因此,有条件的话建议使用优级纯试剂,尽量降低试剂中的含氮量,从而降低实验空白值。5、实验用水及试剂的质量检验[b][/b]若实验的空白值不够理想,则需要对实验用水及试剂进行检验,以选择出含氮量最低的水和试剂,获得理想的空白值。[color=#ac1d10][b]粗蛋白测定空白偏高的影响因素?[/b][/color]可以从以下方面考虑:1.配溶液的水换没换;2.看看消化炉的回收率;3.看看蒸馏仪的回收率;4.盐酸在不在保质期内;5.配溶液的硫酸是不是有问题;6.检查催化剂,是不是含有硝酸钾。小结空白试验是实验室质量控制的重要环节,其准确性对提高检测结果的准确度至关重要。以分光光度法空白为例 ,要用相应的实验用纯水或有机溶济作参比,不能用空白实验溶液做参比,以免人为减小空白值,从而掩盖空白实验值的变异 。值得注意的是,通过计量认证的实验室,由于检测仪器和玻璃量器定期检定 ,以及采取了全面的质量管理措施,因此,若测定的空白实验值太大,一般是化学药品纯度不够,配制的试液变质或被污染等原因 。应重新配制试液 ,返工重测该批样品 ,直至空白实验值合格。
在蜂蜜糖分检测中,使用液相和示差,除了做平行试验还要做空白试验,如何做空白试验???????????[em26]
试验时常看到标准里要求随同试验做空白试验。空白试验是不是这样的:例如称样品1g用浓硝酸10ml溶解、记为1号;而空白试验就是量取10ml浓硝酸、记为2号,以下1号和2号处理方式相同。如果是这样那我有样品需要20ml硝酸消解定溶50ml,那20ml硝酸做空白岂不是太多
试验时常提到做空白试验。空白试验是不是这样的:例如称样品1g用浓硝酸10ml溶解、记为1号;而空白试验就是量取10ml浓硝酸、记为2号,以下1号和2号处理方式相同。如果是这样那我有样品需要20ml硝酸消解定溶50ml,那20ml硝酸做空白岂不是太多?
疑问:是否还需要加入空白试验?一、前提:标曲工作液用浸提剂定容,土壤中加入20毫升浸提剂。二、工作步骤如下。 1、用水或空气调零。 2、做标曲。 3、化验土壤铜的含量。三、工作中的出现的疑问是(随便捏个数字做个比方): 1、看到的吸光值: 吸光值 浓度 0.001 0 0.043 1 0.082 2 0.125 3 0.167 4 0.201 52、当看到上面显示的吸光值时,用鼠标点击后,变成了: 吸光值 浓度 0.001 0 0.042 1 0.081 2 0.124 3 0.166 4 0.200 5 从数字上可看出,电脑自动就扣除了空白的吸光度。3、参予标曲相关系数R的计算的数字是: 吸光值 浓度 0.042 1 0.081 2 0.124 3 0.166 4 0.200 5 即:仅对含铜的标曲工作液进行相关系数R的计算。 请教与讨论的是:做完标曲后,还用再加入空白试验吗?如果加入空白试验,计算化验结果,化验结果则是两次扣除浸提剂吸光度引起的浓度,这样做对吗?4、个人观点,如果加入空白试验,是不对的。只有在用超纯水定容标准工作液的情况下,才可以加入空白试验,消除浸提剂引起的误差。 敬请参加讨论,因为加入空白试验的化验太多,而且有的是直接以标曲空白工作液调零。请各述高见。以更全面的理解标曲与化验的关系。
非水滴定中,空白试验和对照试验用滴定液差不多一样,可能的原因
空白试验到底是用配制标物的试剂,还是用萃取样品的溶剂?这两个我用的是不同的试剂。
实验室内部质控,他要求石墨炉原子吸收光谱法测定镉的空白试验,这个怎么做 然后怎么评价写结论?求指导
原荧中,空白试验是指用纯水代替样品,像样品一样走消解步骤。 那样品空白是什么意思?
按照[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]GB方法检测的,每个标准里都有个方法空白试验,你们觉得有必要做方法空白么?不知道有没有人空白试验做出来有目标物的,是什么组分?
我们经常会做空白试验,想必一定有曾经让你纠结、头疼、费心的空白试验吧,你还记得都是什么吗?说说曾经最让你头疼的空白试验,分享你的空白试验经历和解决办法,就可以获得2-10个积分奖励!!!======================================================================相关话题:1、【盘点分析空白】ICPMS的空白来源与消除http://bbs.instrument.com.cn/shtml/20130203/4555706/2、【盘点分析空白】化学分析的空白分类与意义http://bbs.instrument.com.cn/shtml/20130216/4568655/3、【盘点分析空白】液质联用检测的空白干扰问题http://bbs.instrument.com.cn/shtml/20130219/4573981/4、【盘点分析空白】聊聊你曾经做过的那些试剂空白http://bbs.instrument.com.cn/shtml/20130221/4578495/5、【盘点分析空白】说说曾经最让你头疼的空白试验http://bbs.instrument.com.cn/shtml/20130225/4584924/
质量控制的计划中需要做空白试验,我不懂是什么意思?
土壤元素测定的空白试验怎么做? 是用石英砂做替代样品吗? 我做的是土壤全量钾钠钙镁。土壤有机质的测定里,空白试验是用石英砂替代样品的,其它元素的空白试验在书上都没写啊?是一样用石英砂替代样品吗?求给位前辈多多指教啦!急!急!急!
曾经最让你头疼的空白试验有哪些?就是怎么做也不稳定,跑偏的那种?大家有没?
[size=2]如题,在工作中发现一个问题:药典附录中,硫酸滴定液的标定未规定做空白试验,但在GB/T 601—2002(化学试剂、标准滴定溶液的制备)中,又规定了需做空白试验,实际工作中需要做吗?另外,药典标准的滴定分析方法中,如果要做空白试验的话,一般都说明了.....[/size]

实验的时候,标准要求实验值减去空白试验值。实际中发现,经常有用一个空白试验值或者作一个空白试验的现象。请大家看看,空白试验作单实验还是双实验?以蛋白质测定5009.5第一法为例6.1 凯氏定氮法6.1.1 试样处理:称取充分混匀的固体试样0.2 g~2 g、半固体试样 2 g~5 g 或液体试样10 g~25 g(约相当于30 mg~40 mg 氮),精确至0.001 g,移入干燥的100 mL、250 mL 或500 mL 定氮瓶中,加入.......................放冷后,移入100 mL 容量瓶中,并用少量水洗定氮瓶,洗液并入容量瓶中,再加水至刻度,混匀备用。同时做试剂空白试验。6.1.2 测定:......................以硫酸或盐酸标准滴定溶液(4.12)滴定至终点,其中2 份甲基红乙醇溶液(4.13)与1 份亚甲基蓝乙醇溶液(4.14)指示剂,颜色由紫红色变成灰色,pH 5.4;1 份甲基红乙醇溶液(4.13)与5 份溴甲酚绿乙醇溶液(4.15)指示剂,颜色由酒红色变成绿色,pH 5.1。同时作试剂空白。http://ng1.17img.cn/bbsfiles/images/2012/10/201210311537_400493_1722582_3.jpg
标定盐酸时的空白试验怎么做啊?是不加基准物质碳酸钠,其它步骤不变吗?如果是,我滴定空白试验是怎么一滴后颜色就变成亮粉色了呢?再加热煮沸两分钟后,颜色不但没退反而还加深了,这是为什么?
痕量检测,如黄曲霉素、重金属、农残等有规定必须要每批做空白试验吗?如果做空白试验,怎么做?
6校准曲线的绘制 分别移取0 mL,l. 00 mL,2. 00 mL,3. 00 mL,4. 00 mL及5. 00 mL锌标准溶液于6只50 mL容量瓶中,加人约30 mL水。此系列溶液分别含有0 mg, 0. 01 mg, 0. 02 mg, 0. 03 mg, 0. 04 mg, 0. 05 mg锌。分别加人10. 0 mL硼酸一氯化钾一氢氧化钠缓冲溶液和5. 00 mL锌试剂溶液,用水稀释至刻度,摇匀,放置IO min。于620 nm处,用2 cm吸收池,以试剂空白为参比,测定其吸光度。 以吸光度为纵坐标,锌含量(mg)为横坐标,绘制校准曲线。7. 1含有机麟酸盐的试样的分析步骤 移取适量体积的试样于l00 mL烧杯中,加人0. 5 mL硫酸溶液及1 mL过硫酸按溶液,加水至溶液总体积约为30 mL。在电炉上煮沸5 min,取下,冷却至室温。加人1滴甲基橙指示液,用氢氧化钠溶液调至溶液呈黄色。将溶液全部转移至50 mL容量瓶中,加人10. 0 mL硼酸一氯化钾一氢氧化钠缓冲溶液和5. 00 mL锌试剂溶液,用水稀释至刻度,摇匀,放置10 min。于620 nm处,用2 cm吸收池,以空白试验为参比,测定其吸光度,从校准曲线上查出相应的锌的含量(mg) o7. 2不含有机麟酸盐的试样的分析步骤 移取适量体积的试样,于50 mL容量瓶中,加人30 mL水,以下操作按照第6章中从“加人10. 0 mL硼酸一氯化钾一氢氧化钠缓冲溶液……”开始,进行操作。以空白试验为参比。于620 nm处,测定其吸光度。其中的试剂空白与空白试验应该分别怎样做
现在有些老师讲化验说要做个空白试验,还有一些国家类的书也是这样写的.这个空白不是标曲的空白,而是说我做土壤,做完一个标曲,我也要带进一个只含有浸提剂的空白. 对这种方法我质疑。举例讲我化验土壤中的铜。工作步骤如下。1、用水或空气调零。2、做标曲。3、化验土壤含量。疑问是(随便捏个数字做个比方):1、看到的吸光值:吸光值 浓度 0.001 0 0.043 1 0.082 2 0.125 3 0.167 4 0.201 52、当看到上面显示的吸光值时,用鼠标去点击时,实际则变成了:吸光值 浓度 0.001 0 0.042 1 0.081 2 0.124 3 0.166 4 0.200 5即电脑自动就扣除了空白的吸光度。3、参予标曲工作液相关系数R的计算的数字是: 0.042 1 0.081 2 0.124 3 0.166 4 0.200 5需要注意的是:配制的标准工作液,用浸提剂进行定容,土壤振荡前加入的也是浸提剂。请教与讨论的是:做土壤化验时,也就是做完标曲后,还用再加入空白试验吗?如果加入,计算化验结果,就成了两次扣除浸提剂,这样对吗?
微波消解法空白试验比样品结果高?是消解罐污染吗?
液相色谱的试验中,空白试样的色谱图怎样参与计算,是试样和空白试样的两个色谱图进行相减吗????????????
如题,卡尔费休法测水分应该做几份空白试验?一份足矣么......
[color=#333333]食品分析中什么叫空白试验[/color]
如题 比较清洁的水样,做悬浮物指标时是否需要做空白试验?国标是GB11901-1989 标准上没有明确说明,说的很简单。
在COD测试中,加入HgSO4抗氯化物干扰时,在空白试验时,是否也要加入HgSO4。
急!急!急!GB5413.2010 婴幼儿食品和乳品中脂肪的测定要求做空白试验,6.2写的是水,但9.2要求的是无水奶油,到底用哪一个?原因?
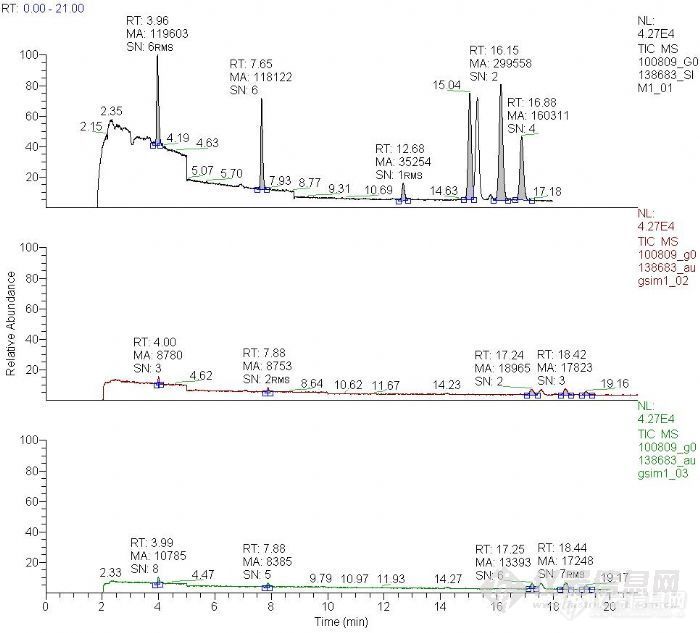
在用PE公司的管子做二次解吸的试验,不过试了很多次,跑管子的第一次时基本上所要的峰都比较高,在同样的条件下紧接着跑第二次第三次时,按说如果二次吸附解吸的比较完全的话,第二次出峰时对应的峰应高很小或者接近没有了吧,也就是说可以当作空白样品,当然也可以以此来确定所设的条件是否合适。对条件已经探索了很久,希望能把第二次的结果当作空白,不过似乎总有点勉强。附上我连续做的三次结果:虽然后面2次的峰面积确实已经小了很多,但始终都有,而且有时信噪比还挺大,至少比3大。请问各位,怎么定义样品的空白呢?就是说在空白试验时,目标化合物的峰高、峰面积或者信噪比应该达到什么值才能不予考虑。能不能在每次计算样品的峰高时减去空白中的峰面积再利用标准曲线算出样品的浓度呢?还有就是第一次跑的样品结果尽管峰看着挺高,但是信噪比为什么那么低呢?跑的时候略微改了一点程序升温,所以保留时间有些差别。[img]http://ng1.17img.cn/bbsfiles/images/2010/08/201008092242_235504_1833417_3.jpg[/img]







 我要推广仪器
我要推广仪器
 下载APP
下载APP