色谱柱如果在样品分析时发现每个色谱峰都有双峰出现,尤其在采用单一纯物质时,则可以确定是色谱柱出现问题了,一般是柱头受损或者柱头固定相污染引起的。如果进样量少,原来色谱柱正常,色谱峰的形状多为一大峰带一小峰,不一定拖尾,这一般应是柱头受堵,将色谱柱反过来接,用流动相冲洗或酸洗或其它溶剂,将堵在柱头的残留物冲掉,再反过来,一般情况下就行了。当然不反冲,正冲有时也会正常的。如果峰拖尾,双峰强弱相差不大,柱头固定相变脏或流失可能性更大,这时可以将进样头拧开,将微孔滤片超声,柱头刮去一部分填料,重新填上新填料拧紧,不过这个对技术要求较高且不能经常做,否则用不了几次,色谱柱就会应柱效降低而报废。如果上述不能解决问题,可能是柱塌陷造成的,此时需要更换色谱柱。溶剂问题目前HPLC分析多为反相色谱,流动相多为甲醇、乙腈、水,加各种添加剂以改善分离性能。样品一般用与流动相相溶的溶剂溶解,最佳的溶解方法是用流动相溶解。在实际分析中,有时候为了样品的溶解性或稳定性加入了一点的缓冲液,缓冲液的酸碱性可能会导致样品转化,从而产生双峰现象。此时需要更换缓冲液,调整溶剂pH值,或者用流动相配置样品。此外,样品要现配现用,避免样品溶解液的有机相比例、pH值发生变化而导致的溶剂效应。进样量当用溶剂极性强度大的试剂,如纯甲醇、纯乙腈,纯乙醇,而分析体系中以水为主时,如果样品进样量大,如定量管为20ul,此时单一的纯物质会出现双峰,第二峰比第一峰小(每次都不太一样),且拖尾,保留时间会提前(相对进样量少而言),将进样量减少一半以上,峰型将变为正常。这是样品的溶剂与流动相极性相差太大,而流动相来不及将其稀释达到平衡造成的,此情况下需要减少进样量。另一个原因是,进样量不一定大,但绝对量很大,色谱图上的双峰紧靠在一起,基本上齐高,不拖尾(如果出峰很快,也可能是色谱柱问题)。将样品稀释再进样就可以了,这是由于进样量过大,色谱柱过载造成的。pH值体系pH值对色谱双峰的影响出现在各个环节上(前面已经提到),尤其在缓冲液流动相平衡过程中其影响更为明显。当连续进样时,受pH的连续变化影响会经常遇到这种双峰的情况。另外,在样品分析时,流动相的pH尽量远离被分析物的等电点,否则也容易引起双峰的产生。在用离子对试剂分析时,选择不好条件也会容易引起双峰的产生。样品特性有些样品由于其化学结构的特点,存在互变异构现象,而这种互变异构体无法分开,而是以一个动态平衡存在。在色谱分析时,在一个特定的条件下(如pH值、流动相极性、温度等),一种物质将出现双峰,双峰靠的很近,基本齐高,不拖尾,条件稍一变化,尤其pH,双峰现象将消失,如红霉素等。有的样品紫外的色谱图上看不到双峰,但在[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]下,用质谱检测器,其质谱的总离子流图上较明显,如农药啶虫眯(吡虫清)。
[font=&][color=#333333]色谱双峰指的是明明是同一种物质,但在色谱图中却呈现双峰,让实验人员误认为含有两种物质。[/color][/font][font=-apple-system, BlinkMacSystemFont, &][color=#333333]那么,出现双峰的原因主要有哪些呢?[/color][/font][font=&][size=14px][b]出现色谱双峰的原因一般有以下几种原因:色谱柱的问题、溶剂、进样量、样品特性,以及pH。[/b][/size][/font][font=&][size=14px][color=#ff4c00][b][/b][/color][/size][/font]色谱柱:[font=&][color=#333333]如果在样品分析时发现每个色谱峰都有双峰出现,尤其在采用单一纯物质时,则可以确定是色谱柱出现问题了,一般是柱头受损或者柱头固定相污染引起的。[font=&]如果进样量少,原来色谱柱正常,色谱峰的形状多为一大峰带一小峰,不一定拖尾,这一般应是柱头受堵,将色谱柱反过来接,用流动相冲洗或酸洗或其它溶剂,将堵在柱头的残留物冲掉,再反过来,一般情况下就行了。当然不反冲,正冲有时也会正常的。[/font][font=&]如果峰拖尾,双峰强弱相差不大,柱头固定相变脏或流失可能性更大,这时可以将进样头拧开,将微孔滤片超声,柱头刮去一部分填料,重新填上新填料拧紧,不过这个对技术要求较高且不能经常做,否则用不了几次,色谱柱就会应柱效降低而报废。[/font][font=&]如果上述不能解决问题,可能是柱塌陷造成的,此时需要更换色谱柱。[/font][/color][/font][color=#333333][font=&][size=14px]溶剂问题:目前HPLC分析多为反相色谱,流动相多为甲醇、乙腈、水,加各种添加剂以改善分离性能。样品一般用与流动相相溶的溶剂溶解,最佳的溶解方法是用流动相溶解。在实际分析中,有时候为了样品的溶解性或稳定性加入了一点的缓冲液,缓冲液的酸碱性可能会导致样品转化,从而产生双峰现象。此时需要更换缓冲液,调整溶剂pH值,或者用流动相配置样品。此外,样品要现配现用,避免样品溶解液的有机相比例、pH值发生变化而导致的溶剂效应。[/size][/font][/color][color=#333333][font=&][size=14px]进样量:当用溶剂极性强度大的试剂,如纯甲醇、纯乙腈,纯乙醇,而分析体系中以水为主时,如果样品进样量大,如定量管为20ul,此时单一的纯物质会出现双峰,第二峰比第一峰小(每次都不太一样),且拖尾,保留时间会提前(相对进样量少而言),将进样量减少一半以上,峰型将变为正常。这是样品的溶剂与流动相极性相差太大,而流动相来不及将其稀释达到平衡造成的,此情况下需要减少进样量。另一个原因是,进样量不一定大,但绝对量很大,色谱图上的双峰紧靠在一起,基本上齐高,不拖尾(如果出峰很快,也可能是色谱柱问题)。将样品稀释再进样就可以了,这是由于进样量过大,色谱柱过载造成的。[/size][/font][/color][size=14px][font=&][size=14px]样品特性:有些样品由于其化学结构的特点,存在互变异构现象,而这种互变异构体无法分开,而是以一个动态平衡存在。在色谱分析时,在一个特定的条件下(如pH值、流动相极性、温度等),一种物质将出现双峰,双峰靠的很近,基本齐高,不拖尾,条件稍一变化,尤其pH,双峰现象将消失,如红霉素等。有的样品紫外的色谱图上看不到双峰,但在[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]下,用质谱检测器,其质谱的总离子流图上较明显,如农药啶虫眯(吡虫清)。[/size][/font][/size][size=14px][size=14px][color=#333333]PH值:体系pH值对色谱双峰的影响出现在各个环节上(前面已经提到),尤其在缓冲液流动相平衡过程中其影响更为明显。当连续进样时,受pH的连续变化影响会经常遇到这种双峰的情况。另外,在样品分析时,流动相的pH尽量远离被分析物的等电点,否则也容易引起双峰的产生。在用离子对试剂分析时,选择不好条件也会容易引起双峰的产生。[/color][/size][/size][font=&][font=&][size=14px][/size][/font][/font][color=#333333][font=&][color=#333333][/color][/font][/color][font=-apple-system, BlinkMacSystemFont, &][color=#333333][/color][/font]
色谱双峰产生的可能及判断和处理 HPLC分析中,在色谱柱正常,样品灵敏度足够,分析方法合适,色谱峰在出峰时间较短的条件下(不包括梯度),峰型应对称而尖锐。但是,在对样品了解程度不够,方法不妥,样品处理方法及进样方式不合理下,会出现各种意想不到的问题,而对色谱峰难以作出合理的解释,尤其对于新手更是如此。 色谱双峰指的是不是一种物质,但在色谱图中出现双峰,而表明含二种物质。可将这种情况分为三种原因。1 色谱柱如果你分析样品时发现每个色谱峰都双峰出现(出峰越快,双峰的可能性会减少),尤其采用单一纯物质时,可以肯定色谱柱出问题-柱头受损或柱头固定相变脏或流失。如果进样量少,原来色谱柱正常,色谱峰的形状多为一大峰带一小峰,不一定拖尾,这一般应是柱头受堵,将色谱柱反过来接,用流动相冲洗或酸洗或其它溶剂,将堵在柱头的残留物冲掉,再反过来,一般情况下就行了。当然不反冲,正冲有时也会正常的。如果峰拖尾,双峰强弱相差不大,柱头固定相变脏或流失可能性更大,这是可以将进样那头拧开,将微孔滤片超声,柱头刮去一部分填料,重新填上新填料拧紧,不过这种活,需要一定技术,同时不能老干那种事,否则用不了几次,色谱柱就会应柱效很低而报废。2 溶剂极性及进样量许多HPLC分析者对此可能不以为然,一般的HPLC的书籍和文献都不会提到这方面的内容,而这确是双峰产生的一个很重要的原因。目前HPLC分析多为反相色谱,流动相多为甲醇、乙腈、水,加各种添加剂以改善分离性能。样品一般用与流动相相溶的溶剂溶解。最佳的溶解方法是用流动相溶解,但是很多情况是不一致的。当用溶剂极性强度大的试剂,如纯甲醇、纯乙腈,纯乙醇,而分析体系中以水为主,样品进样量大,如定量管为20ul,此条件下完全可以肯定,单一的纯物质出双峰,第二峰比第一峰小(每次都不太一样),且拖尾,保留时间会提前(相对进样量少而言),将进样量减少一半以上,峰型将变为正常。这是样品的溶剂与流动相极性相差太大,而流动相来不及将其稀释达到平衡造成的。上面提到进样量造成双峰的一个原因,另一个原因是,进样量不一定大,但绝对量很大,色谱图上的双峰紧靠在一起,基本上齐高,不拖尾(如果出峰很快,也可能是色谱柱问题)。将样品稀释再进样就可以了,这是由于进样量过大,色谱柱过载造成的。3 样品的特性有些样品由于其化学结构的特点,存在互变异构现象,而这种互变异构体无法分开,而是以一个动态平衡存在。在色谱分析时,在一个特定的条件下,一种物质将出现双峰,双峰靠的很近,基本齐高,不拖尾,条件稍一变化,尤其pH,双峰现象将消失,如红霉素等。有的样品紫外的色谱图上看不到双峰,但在LC-MS下,用质谱检测器,其质谱的总离子流图上较明显,例如农药啶虫眯(吡虫清)。
色谱双峰产生的可能及判断和处理HPLC分析中,在色谱柱正常,样品灵敏度足够,分析方法合适,色谱峰在出峰时间较短的条件下(不包括梯度),峰型应对称而尖锐。但是,在对样品了解程度不够,方法不妥,样品处理方法及进样方式不合理下,会出现各种意想不到的问题,而对色谱峰难以作出合理的解释,尤其对于新手更是如此。色谱双峰指的是不是一种物质,但在色谱图中出现双峰,而表明含二种物质。可将这种情况分为三种原因。1 色谱柱如果你分析样品时发现每个色谱峰都双峰出现(出峰越快,双峰的可能性会减少),尤其采用单一纯物质时,可以肯定色谱柱出问题-柱头受损或柱头固定相变脏或流失。如果进样量少,原来色谱柱正常,色谱峰的形状多为一大峰带一小峰,不一定拖尾,这一般应是柱头受堵,将色谱柱反过来接,用流动相冲洗或酸洗或其它溶剂,将堵在柱头的残留物冲掉,再反过来,一般情况下就行了。当然不反冲,正冲有时也会正常的。如果峰拖尾,双峰强弱相差不大,柱头固定相变脏或流失可能性更大,这是可以将进样那头拧开,将微孔滤片超声,柱头刮去一部分填料,重新填上新填料拧紧,不过这种活,需要一定技术,同时不能老干那种事,否则用不了几次,色谱柱就会应柱效很低而报废。2 溶剂极性及进样量许多HPLC分析者对此可能不以为然,一般的HPLC的书籍和文献都不会提到这方面的内容,而这确是双峰产生的一个很重要的原因。目前HPLC分析多为反相色谱,流动相多为甲醇、乙腈、水,加各种添加剂以改善分离性能。样品一般用与流动相相溶的溶剂溶解。最佳的溶解方法是用流动相溶解,但是很多情况是不一致的。当用溶剂极性强度大的试剂,如纯甲醇、纯乙腈,纯乙醇,而分析体系中以水为主,样品进样量大,如定量管为20ul,此条件下完全可以肯定,单一的纯物质出双峰,第二峰比第一峰小(每次都不太一样),且拖尾,保留时间会提前(相对进样量少而言),将进样量减少一半以上,峰型将变为正常。这是样品的溶剂与流动相极性相差太大,而流动相来不及将其稀释达到平衡造成的。上面提到进样量造成双峰的一个原因,另一个原因是,进样量不一定大,但绝对量很大,色谱图上的双峰紧靠在一起,基本上齐高,不拖尾(如果出峰很快,也可能是色谱柱问题)。将样品稀释再进样就可以了,这是由于进样量过大,色谱柱过载造成的。3 样品的特性有些样品由于其化学结构的特点,存在互变异构现象,而这种互变异构体无法分开,而是以一个动态平衡存在。在色谱分析时,在一个特定的条件下,一种物质将出现双峰,双峰靠的很近,基本齐高,不拖尾,条件稍一变化,尤其pH,双峰现象将消失,如红霉素等。有的样品紫外的色谱图上看不到双峰,但在LC-MS下,用质谱检测器,其质谱的总离子流图上较明显,例如农药啶虫眯(吡虫清)。
hplc分析中,在色谱柱正常,样品灵敏度足够,分析方法合适,色谱峰在出峰时间较短的条件下(不包括梯度),峰型应对称而尖锐。但是,在对样品了解程度不够,方法不妥,样品处理方法及进样方式不合理下,会出现各种意想不到的问题,而对色谱峰难以作出合理的解释,尤其对于新手更是如此。下面根据本人几年工作的体会,提出一些看法,向同仁指教。 色谱双峰指的是明是一种物质,但在色谱图中出现双峰,而表明含二种物质。我将这种情况分为四种原因。 1 色谱柱 如果你分析样品时发现每个色谱峰都双峰出现(出峰越快,双峰的可能性会减少),尤其采用单一纯物质时,可以肯定色谱柱出问题-柱头受损或柱头固定相变脏或流失。如果进样量少,原来色谱柱正常,色谱峰的形状多为一大峰带一小峰,不一定拖尾,这一般应是柱头受堵,将色谱柱反过来接,用流动相冲洗或酸洗或其它溶剂,将堵在柱头的残留物冲掉,再反过来,一般情况下就行了。当然不反冲,正冲有时也会正常的。如果峰拖尾,双峰强弱相差不大,柱头固定相变脏或流失可能性更大,这是可以将进样那头拧开,将微孔滤片超声,柱头刮去一部分填料,重新填上新填料拧紧,不过这种活,需要一定技术,同时不能老干那种事,否则用不了几次,色谱柱就会应柱效很低而报废。2 溶剂极性及进样量 许多hplc分析者对此可能不以为然,一般的hplc的书籍和文献都不会提到这方面的内容,而这确是双峰产生的一个很重要的原因。目前hplc分析多为反相色谱,流动相多为甲醇、乙腈、水,加各种添加剂以改善分离性能。样品一般用与流动相相溶的溶剂溶解。最佳的溶解方法是用流动相溶解,但是很多情况是不一致的。当用溶剂极性强度大的试剂,如纯甲醇、纯乙腈,纯乙醇,而分析体系中以水为主,样品进样量大,如定量管为20ul,此条件下完全可以肯定,单一的纯物质出双峰,第二峰比第一峰小(每次都不太一样),且拖尾,保留时间会提前(相对进样量少而言),将进样量减少一半以上,峰型将变为正常。这是样品的溶剂与流动相极性相差太大,而流动相来不及将其稀释达到平衡造成的。上面提到进样量造成双峰的一个原因,另一个原因是,进样量不一定大,但绝对量很大,色谱图上的双峰紧靠在一起,基本上齐高,不拖尾(如果出峰很快,也可能是色谱柱问题)。将样品稀释再进样就可以了,这是由于进样量过大,色谱柱过载造成的。 3 样品的特性 有些样品由于其化学结构的特点,存在互变异构现象,而这种互变异构体无法分开,而是以一个动态平衡存在。在色谱分析时,在一个特定的条件下,一种物质将出现双峰,双峰靠的很近,基本齐高,不拖尾,条件稍一变化,尤其ph,双峰现象将消失,如红霉素等。有的样品紫外的色谱图上看不到双峰,但在lc-ms下,用质谱检测器,其质谱的总离子流图上较明显,例如我分析过的农药啶虫眯(吡虫清)。4 参数 记录的参数一般都内定的,不必修改,但gc和hplc的参数是不完全一致的,例如c-r3a数据记录仪上的一般记录时间间隔gc为2ms,hplc 为5ms,如果记录间隔时间缩短,一个峰将变为二个峰或更多。 --转自中国色谱网
[b][font=微软雅黑, &][color=#333333]出现色谱双峰的原因一般有以下几种原因:色谱柱的问题、溶剂、进样量、样品特性,以及pH。[/color][/font][/b][align=center][b][font=微软雅黑, &][color=#333333][b][size=16px]色谱柱[/size][/b][/color][/font][/b][/align][b][font=微软雅黑, &][b][size=16px][color=#333333]1、如果在样品分析时发现[b]每个色谱峰都有双峰出现[/b],尤其在采用单一纯物质时,则可以确定是色谱柱出现问题了,一般是[b]柱头受损或者柱头固定相污染引起的。[/b]2、[b]如果进样量少,原来色谱柱正常,色谱峰的形状多为一大峰带一小峰,不一定拖尾,这一般应是柱头受堵,[/b]将色谱柱反过来接,适当溶剂冲洗,将堵在柱头的残留物冲掉,再反过来,一般情况下就行了。当然不反冲,正冲有时也会正常的。3、[b]如果峰拖尾,双峰强弱相差不大,柱头固定相变脏或流失可能性更大,[/b]这时可以将进样头拧开,将微孔滤片超声,柱头刮去一部分填料,重新填上新填料拧紧,不过这个对技术要求较高且不能经常做,否则用不了几次,色谱柱就会应柱效降低而报废。4、如果上述不能解决问题,可能是柱塌陷造成的,此时需要更换色谱柱。[/color][/size][/b][/font][/b][align=center][b][font=微软雅黑, &][b][size=16px][font=微软雅黑, &][b]溶剂问题[/b][/font][/size][/b][/font][/b][/align][b][font=微软雅黑, &][b][size=16px][font=微软雅黑, &][b]1、目前HPLC分析多为反相色谱,流动相多为甲醇、乙腈、水,加各种添加剂以改善分离性能。样品一般用与流动相相溶的溶剂溶解,最佳的溶解方法是用流动相溶解。在实际分析中,有时候为了样品的溶解性或稳定性加入了一点的缓冲液,缓冲液的酸碱性可能会导致样品转化,从而产生双峰现象。此时[b]需要更换缓冲液,调整溶剂pH值,或者用流动相配置样品。[/b]2、此外,样品要现配现用,避免样品溶解液的有机相比例、pH值发生变化而导致的溶剂效应。[/b][/font][/size][/b][/font][/b][align=center][b][b][font=微软雅黑, &][size=14px][b][font=微软雅黑, &][b]进样量[/b][/font][/b][/size][/font][/b][/b][/align][b][b][font=微软雅黑, &][b][font=微软雅黑, &][size=14px][b]1、[b]当用溶剂极性强度大的试剂,如纯甲醇、纯乙腈,纯乙醇,而分析体系中以水为主时,如果样品进样量大,如定量管为20ul,此时单一的纯物质会出现双峰,第二峰比第一峰小(每次都不太一样),且拖尾,保留时间会提前(相对进样量少而言),将进样量减少一半以上,峰型将变为正常。这是样品的溶剂与流动相极性相差太大,而流动相来不及将其稀释达到平衡造成的,此情况下需要减少进样量。[/b]2、另一个原因是,进样量不一定大,但绝对量很大,色谱图上的双峰紧靠在一起,基本上齐高,不拖尾(如果出峰很快,也可能是色谱柱问题)。将样品稀释再进样就可以了,[b]这是由于进样量过大,色谱柱过载造成的。[/b][/b][/size][/font][/b][/font][/b][/b][align=center][b][font=微软雅黑, &][b][font=微软雅黑, &][b][font=微软雅黑, &][b][b][b]样品特性[/b][/b][/b][/font][/b][/font][/b][/font][/b][/align][b][b][b][font=微软雅黑, &][b][size=16px][font=微软雅黑, &][b][b][b]有些样品由于其化学结构的特点,存在互变异构现象,而这种互变异构体无法分开,而是以一个动态平衡存在。[/b]在色谱分析时,在一个特定的条件下(如pH值、流动相极性、温度等),一种物质将出现双峰,双峰靠的很近,基本齐高,不拖尾,条件稍一变化,尤其pH,双峰现象将消失,如红霉素等。有的样品紫外的色谱图上看不到双峰,但在[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]下,用质谱检测器,其质谱的总离子流图上较明显,如农药啶虫眯(吡虫清)。[/b][/b][/font][/size][/b][/font][/b][/b][/b][align=center][b][b][font=微软雅黑, &][b][font=微软雅黑, &][b][size=16px][b][b][b]pH值[/b][/b][/b][/size][/b][/font][/b][/font][/b][/b][/align][b][b][b][font=微软雅黑, &][b][font=微软雅黑, &][b][b][size=14px][b][b]体系pH值对色谱双峰的影响出现在各个环节上(前面已经提到),尤其在缓冲液流动相平衡过程中其影响更为明显。[/b]当连续进样时,受pH的连续变化影响会经常遇到这种双峰的情况。另外,在样品分析时,流动相的pH尽量远离被分析物的等电点,否则也容易引起双峰的产生。在用离子对试剂分析时,选择不好条件也会容易引起双峰的产生。[/b][/size][/b][/b][/font][/b][/font][/b][b][b][size=14px][b][b][font=微软雅黑, &][size=14px][b]HPLC分析中,在色谱柱正常,样品灵敏度足够,分析方法合适,色谱峰在出峰时间较短的条件下(不包括梯度),峰型应对称而尖锐。但是,在对样品了解程度不够,方法不妥,样品处理方法及进样方式不合理下,会出现各种意想不到的问题,而对色谱峰难以作出合理的解释,尤其对于新手更是如此,所以,了解每种鬼峰出现的原因对我们的分析工作是非常重要的。[/b][/size][/font][/b][/b][/size][/b][/b][b][b][size=14px][b][b][font=微软雅黑, &][size=14px][b][/b][/size][/font][/b][/b][/size][/b][/b][/b][/b]
HPLC分析中,在色谱柱正常,样品灵敏度足够,分析方法合适,色谱峰在出峰时间较短的条件下(不包括梯度),峰型应对称而尖锐。但是,在对样品了解程度不够,方法不妥,样品处理方法及进样方式不合理下,会出现各种意想不到的问题,而对色谱峰难以作出合理的解释,尤其对于新手更是如此。下面根据本人几年工作的体会,提出一些看法,向同仁指教。 色谱双峰指的是明是一种物质,但在色谱图中出现双峰,而表明含二种物质。我将这种情况分为四种原因。 1 色谱柱 如果你分析样品时发现每个色谱峰都双峰出现(出峰越快,双峰的可能性会减少),尤其采用单一纯物质时,可以肯定色谱柱出问题-柱头受损或柱头固定相变脏或流失。如果进样量少,原来色谱柱正常,色谱峰的形状多为一大峰带一小峰,不一定拖尾,这一般应是柱头受堵,将色谱柱反过来接,用流动相冲洗或酸洗或其它溶剂,将堵在柱头的残留物冲掉,再反过来,一般情况下就行了。当然不反冲,正冲有时也会正常的。如果峰拖尾,双峰强弱相差不大,柱头固定相变脏或流失可能性更大,这是可以将进样那头拧开,将微孔滤片超声,柱头刮去一部分填料,重新填上新填料拧紧,不过这种活,需要一定技术,同时不能老干那种事,否则用不了几次,色谱柱就会应柱效很低而报废。 2 溶剂极性及进样量 许多HPLC分析者对此可能不以为然,一般的HPLC的书籍和文献都不会提到这方面的内容,而这确是双峰产生的一个很重要的原因。目前HPLC分析多为反相色谱,流动相多为甲醇、乙腈、水,加各种添加剂以改善分离性能。样品一般用与流动相相溶的溶剂溶解。最佳的溶解方法是用流动相溶解,但是很多情况是不一致的。当用溶剂极性强度大的试剂,如纯甲醇、纯乙腈,纯乙醇,而分析体系中以水为主,样品进样量大,如定量管为20ul,此条件下完全可以肯定,单一的纯物质出双峰,第二峰比第一峰小(每次都不太一样),且拖尾,保留时间会提前(相对进样量少而言),将进样量减少一半以上,峰型将变为正常。这是样品的溶剂与流动相极性相差太大,而流动相来不及将其稀释达到平衡造成的。上面提到进样量造成双峰的一个原因,另一个原因是,进样量不一定大,但绝对量很大,色谱图上的双峰紧靠在一起,基本上齐高,不拖尾(如果出峰很快,也可能是色谱柱问题)。将样品稀释再进样就可以了,这是由于进样量过大,色谱柱过载造成的。 3 样品的特性 有些样品由于其化学结构的特点,存在互变异构现象,而这种互变异构体无法分开,而是以一个动态平衡存在。在色谱分析时,在一个特定的条件下,一种物质将出现双峰,双峰靠的很近,基本齐高,不拖尾,条件稍一变化,尤其pH,双峰现象将消失,如红霉素等。有的样品紫外的色谱图上看不到双峰,但在LC-MS下,用质谱检测器,其质谱的总离子流图上较明显,例如我分析过的农药啶虫眯(吡虫清)。 4 参数[
HPLC分析中,在色谱柱正常,样品灵敏度足够,分析方法合适,色谱峰在出峰时间较短的条件下(不包括梯度),峰型应对称而尖锐。但是,在对样品了解程度不够,方法不妥,样品处理方法及进样方式不合理下,会出现各种意想不到的问题,而对色谱峰难以作出合理的解释,尤其对于新手更是如此。下面根据本人几年工作的体会,提出一些看法,向同仁指教。 色谱双峰指的是明是一种物质,但在色谱图中出现双峰,而表明含二种物质。我将这种情况分为四种原因。 1 色谱柱 如果你分析样品时发现每个色谱峰都双峰出现(出峰越快,双峰的可能性会减少),尤其采用单一纯物质时,可以肯定色谱柱出问题-柱头受损或柱头固定相变脏或流失。如果进样量少,原来色谱柱正常,色谱峰的形状多为一大峰带一小峰,不一定拖尾,这一般应是柱头受堵,将色谱柱反过来接,用流动相冲洗或酸洗或其它溶剂,将堵在柱头的残留物冲掉,再反过来,一般情况下就行了。当然不反冲,正冲有时也会正常的。如果峰拖尾,双峰强弱相差不大,柱头固定相变脏或流失可能性更大,这是可以将进样那头拧开,将微孔滤片超声,柱头刮去一部分填料,重新填上新填料拧紧,不过这种活,需要一定技术,同时不能老干那种事,否则用不了几次,色谱柱就会应柱效很低而报废。 2 溶剂极性及进样量 许多HPLC分析者对此可能不以为然,一般的HPLC的书籍和文献都不会提到这方面的内容,而这确是双峰产生的一个很重要的原因。目前HPLC分析多为反相色谱,流动相多为甲醇、乙腈、水,加各种添加剂以改善分离性能。样品一般用与流动相相溶的溶剂溶解。最佳的溶解方法是用流动相溶解,但是很多情况是不一致的。当用溶剂极性强度大的试剂,如纯甲醇、纯乙腈,纯乙醇,而分析体系中以水为主,样品进样量大,如定量管为20ul,此条件下完全可以肯定,单一的纯物质出双峰,第二峰比第一峰小(每次都不太一样),且拖尾,保留时间会提前(相对进样量少而言),将进样量减少一半以上,峰型将变为正常。这是样品的溶剂与流动相极性相差太大,而流动相来不及将其稀释达到平衡造成的。上面提到进样量造成双峰的一个原因,另一个原因是,进样量不一定大,但绝对量很大,色谱图上的双峰紧靠在一起,基本上齐高,不拖尾(如果出峰很快,也可能是色谱柱问题)。将样品稀释再进样就可以了,这是由于进样量过大,色谱柱过载造成的。 3 样品的特性 有些样品由于其化学结构的特点,存
[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LCMS[/color][/url]MS仪器色谱峰出现双峰,峰形变宽,那位老师解答一下影响色谱峰变宽和出双峰的因素?谢谢!出现这种现象的情况:仪器在排了40针运行过程中,前10多针都是好的,而后面20多针就出现峰形变宽和双峰现象。
这篇文章原来早在2001年写成的,但只写了第一部分,刊登在中国色谱网(www.sepu.net)上,但中国色谱网后来几次变局,把文章弄丢了,几年下来,到处转载,网络上漫天飞,大家也不知真的作者是谁,结果变成了网上收集,今天我再次从网上搜到原来的文章,以现在的观点对内容重新修改后刊登在色谱博客网上(www.sepublog.com)。关于第二部分的内容7年后来完成它。 HPLC分析中,在色谱柱正常,样品灵敏度足够,分析方法合适,色谱峰在出峰时间较短的条件下(不包括梯度),峰型应对称而尖锐。但是,在对样品了解程度不够,方法不妥,样品处理方法及进样方式不合理下,会出现各种意想不到的问题,而对色谱峰难以作出合理的解释,尤其对于新手更是如此。下面根据本人几年工作的体会,提出一些看法,向同仁指教。 色谱双峰指的是明明是同一种物质,但在色谱图中出现双峰,而表明含二种物质。我将这种情况分为四种原因。1 色谱柱 如果你分析样品时发现每个色谱峰都双峰出现(出峰越快,双峰的可能性会减少),尤其采用单一纯物质时,可以肯定色谱柱出问题-[color=#DC143C]保护柱污染[/color]或柱头固定相变脏或流失,或[color=#DC143C]颗粒聚集在色谱柱的入口筛板上[/color]。如果进样量少,原来色谱柱正常,色谱峰的形状多为一大峰带一小峰,不一定拖尾,这一般应是柱头受堵,将色谱柱反过来接,用流动相冲洗或酸洗或其它溶剂,将堵在柱头的残留物冲掉,再反过来,一般情况下就行了。当然不反冲,正冲有时也会正常的。 如果峰拖尾,双峰强弱相差不大,柱头固定相变脏或流失可能性更大,这是可以将进样那头拧开,将微孔滤片超声,柱头刮去一部分填料,重新填上新填料拧紧,不过这种活,需要一定技术,同时不能老干那种事,否则用不了几次,色谱柱就会应柱效很低而报废。 2 溶剂极性及进样量 许多HPLC分析者对此可能不以为然,一般的hplc的书籍和文献都不会提到这方面的内容,而这确是双峰产生的一个很重要的原因。目前hplc分析多为反相色谱,流动相多为甲醇、乙腈、水,加各种添加剂以改善分离性能。样品一般用与流动相相溶的溶剂溶解。最佳的溶解方法是用流动相溶解,但是很多情况是不一致的。当用溶剂极性强度大的试剂,如纯甲醇、纯乙腈,纯乙醇,而分析体系中以水为主,样品进样量大,如定量管为20ul,此条件下完全可以肯定,单一的纯物质出双峰,第二峰比第一峰小(每次都不太一样),且拖尾,保留时间会提前(相对进样量少而言),将进样量减少一半以上,峰型将变为正常。这是样品的溶剂与流动相极性相差太大,而流动相来不及将其稀释达到平衡造成的。这是上面提到进样量造成双峰的一个原因,另一个原因是,进样量不一定大,但绝对量很大,色谱图上的双峰紧靠在一起,基本上齐高,不拖尾(如果出峰很快,也可能是色谱柱问题)。将样品稀释再进样就可以了,这是由于进样量过大,色谱柱过载造成的。 [color=#DC143C]如果样品溶剂与流动相不溶或互溶性不好,当样品注入色谱分离体系中,会析出并接着再溶解的可能,这时相当于二次进样,也非常容易产生双峰。[/color].3 样品的特性及pH值 有些样品由于其化学结构的特点,存在互变异构现象,而这种互变异构体无法分开,而是以一个动态平衡存在。在色谱分析时,在一个特定的条件下,一种物质将出现双峰[color=#DC143C],甚至三峰[/color].这时一般双峰靠的很近,基本齐高,不拖尾,条件稍一变化,尤其pH,双峰现象将消失,如红霉素等。有的样品紫外的色谱图上看不到双峰,但在[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]下,用质谱检测器,其质谱的总离子流图上较明显,例如我分析过的农药啶虫眯(吡虫清)。 [color=#DC143C]pH对峰形的影响在缓冲液流动相平衡过程中非常明显,当连续进样时,受pH的连续变化影响会经常遇到这种双峰的情况。另外,在样品分析时,流动相的pH尽量远离被分析物的等电点,否则也容易引起双峰的产生。在用离子对试剂分析时,选择不好条件也会容易引起双峰的产生。 一个pH影响的特例可看我的博客http://sepublog.com/blog/index.php?uid-3-action-viewspace-itemid-143,是关于间苯二甲酸的双峰例子。[/color]4 仪器参数 记录的参数一般都内定的,不必修改,但GC和HPLC的参数是不完全一致的,例如C-R3A数据记录仪上的一般记录时间间隔GC为2ms,HPLC 为5ms,如果记录间隔时间缩短,一个峰将变为二个峰或更多。[color=#DC143C] 还有一种情况是,参比波长设置错误,例如设置分析波长254nm,参比波长400nm,这个对于大多数化合物可能没影响。但是如果被测化合物,在400nm处也有强的紫外吸收,比254nm更高。这样其出峰时,由于背景的抵扣作用,本来一个峰会变成对称的二个峰,而且如果将二峰之间的峰谷反转180度,恰好是一个完整的峰。这时要将参比波长设置更大,或者取消。(红色的为这次新修改的部分)[/color]
当用[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]溶剂极性强度大的试剂,如纯甲醇、纯乙腈,纯乙醇,而分析体系中以水为主时,如果样品进样量大,如定量管为20ul,此时单一的纯物质会出现双峰,第二峰比第一峰小(每次都不太一样),且拖尾,保留时间会提前(相对进样量少而言),将进样量减少一半以上,峰型将变为正常。这是样品的溶剂与流动相极性相差太大,而流动相来不及将其稀释达到平衡造成的,此情况下需要减少进样量。另一个原因是,进样量不一定大,但绝对量很大,色谱图上的双峰紧靠在一起,基本上齐高,不拖尾(如果出峰很快,也可能是色谱柱问题)。将样品稀释再进样就可以了,这是由于进样量过大,色谱柱过载造成的。
求教:菊酯在出双峰是如何进行定量的?软件上如何操作?
在线色谱仪TCD,单峰变双峰,什么原因?
如果在[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]样品分析时发现每个色谱峰都有双峰出现,尤其在采用单一纯物质时,则可以确定是色谱柱出现问题了,一般是柱头受损或者柱头固定相污染引起的。如果进样量少,原来色谱柱正常,色谱峰的形状多为一大峰带一小峰,不一定拖尾,这一般应是柱头受堵,将色谱柱反过来接,用流动相冲洗或酸洗或其它溶剂,将堵在柱头的残留物冲掉,再反过来,一般情况下就行了。当然不反冲,正冲有时也会正常的。如果峰拖尾,双峰强弱相差不大,柱头固定相变脏或流失可能性更大,这时可以将进样头拧开,将微孔滤片超声,柱头刮去一部分填料,重新填上新填料拧紧,不过这个对技术要求较高且不能经常做,否则用不了几次,色谱柱就会应柱效降低而报废。如果上述不能解决问题,可能是柱塌陷造成的,此时需要更换色谱柱。
"问:液相色谱中峰出现拖尾或出现双峰的原因是什么?答: 1. 筛板堵塞或柱失效,解决办法是反向冲洗柱子,替换筛板或更换柱子。2. 存在干扰峰,解决办法为使用较长的柱子,改换流动相或更换选择性好的柱子"液相色谱出现双峰除了上述两个原因还有其他吗?比如说样品浓度过高!因为我将样品浓度再稀释以后,进样检测得到的图谱就不再出现双峰了。[em0808]
[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]体系pH值对色谱双峰的影响在缓冲液流动相平衡过程中其影响更为明显。当连续进样时,受pH的连续变化影响会经常遇到这种双峰的情况。另外,在样品分析时,流动相的pH尽量远离被分析物的等电点,否则也容易引起双峰的产生。在用离子对试剂分析时,选择不好条件也会容易引起双峰的产生。
最近[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]GC2010做车间空气中DMF(二甲基甲酰胺)老出项双峰甚至三峰,峰型很差,色谱条件和以前是一样的:自动进样2微升。进样口温度200摄氏度,压力110千帕。总流量27毫升每分钟,柱流量1.35毫升每分钟,分流比1:20。色谱柱DB-WAX,长30米,膜厚0.25微米,内径0.25毫米。采集时间5.5分钟。色谱温度设置80摄氏度保持1分钟,以10摄氏度每分钟升至100摄氏度,再以20摄氏度每分钟升至150摄氏度。检测器FID,温度250摄氏度。空气流量400毫升每分钟,氢气流量40毫升每分钟。出项双峰可能是什么原因造成的啊?可以通过什么方法排除?
大神们,帮帮忙,刚买的液相Waters色谱柱, 第一次样品出平头峰,第二次用出箭头峰,第三次出现对称的双峰,清洗仪器进样口峰形变丑,是色谱柱哪里污染了吗?仪器污染?还是样品浓度问题?清洗色谱柱第二天还是出现双峰啊,怎么办啊~[img]http://ng1.17img.cn/bbsfiles/images/2018/06/201806091044359237_3688_3364370_3.jpeg[/img][img]http://ng1.17img.cn/bbsfiles/images/2018/06/201806091044365727_6559_3364370_3.jpeg[/img][img]http://ng1.17img.cn/bbsfiles/images/2018/06/201806091044365977_2623_3364370_3.jpeg[/img][img]http://ng1.17img.cn/bbsfiles/images/2018/06/201806091044365979_3071_3364370_3.jpeg[/img]
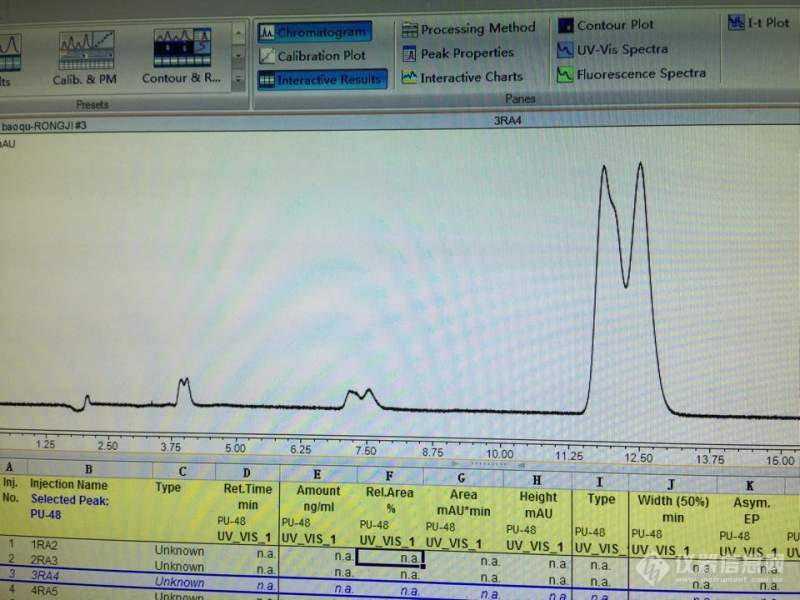
[color=#444444]今天用液相色谱分析,发现实验结果全是双峰,如图所示,保留时间正常,但是色谱峰全都重叠了。请问这是怎么回事,应该怎么办?[/color][color=#444444][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/09/201909021019466880_124_1752329_3.png!w690x517.jpg[/img][/color]
用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定CO2发现出现双峰,这两个双峰是否都应算是CO2的?如何处理.谢谢
[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]有些样品由于其化学结构的特点,存在互变异构现象,而这种互变异构体无法分开,而是以一个动态平衡存在。在色谱分析时,在一个特定的条件下(如pH值、流动相极性、温度等),一种物质将出现双峰,双峰靠的很近,基本齐高,不拖尾,条件稍一变化,尤其pH,双峰现象将消失,如红霉素等。有的样品紫外的色谱图上看不到双峰,但在[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]下,用质谱检测器,其质谱的总离子流图上较明显,如农药啶虫眯(吡虫清)。
液相色谱中峰出现拖尾或出现双峰的原因是什么?
液相色谱中峰出现拖尾或出现双峰的原因是什么?
最近有客户向我们咨询,液相色谱出现连着双峰,是一个还是2个,如何判断?以下解答分享给各位版友,希望对大家有帮助http://simg.instrument.com.cn/bbs/images/default/em09505.gif问:液相色谱出现连着双峰,是一个还是2个,如何判断?答:连着的双峰可能是两种情况导致的: 1、难以分离的两种物质;2、一种物质,由于拖尾严重以至于出现肩峰; 判断方法:用多组分标液进行测试,如果每个主峰后均有一个小峰,则很有可能是肩峰;或者用厂家指定的测试液测试柱性能,如果测试物分叉,则可判断分析时连着的双峰是一种物质分叉所致。
戴安[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]ICS1500,所有峰都是双峰,取下保护柱,单用分离柱正常,于是买了新的保护柱,跑了两个样,一个空白一个混标,正常,第三个混标就又出现峰分叉,咨询热线,说分离柱也有问题,拆下保护柱,单用分离柱跑了10个混标和4个空白,都是正常,请各位大神帮忙分析问题出在哪里?
各位高手请教,岛津2010气相色谱ecd检测器,溶剂怎么会出现双峰? 条件气化室280度,柱箱100度保持2分钟以15度每分钟升到250保持7分钟,检测器温度280,电流1.0 尾吹30.我进了个乙体666,发现溶剂峰出了双峰。
液相色谱中峰出现拖尾或出现双峰的原因是什么?
利用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]检测高纯氢的时候发现了双峰现象,个人认为所用的高纯氢的纯度应该没有问题,但是单个气体为什么总会出现双峰,两个峰的强度也相差不多。当分析氢和氘的混合气体的时候,则是三个峰。为什么会出现这种情况,请各位有经验的朋友给帮忙分析一下,并提供以下解决方案。这里先谢谢大家了。
[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]本来就因为洗脱不到位偶尔出尖峰,拖尾。这次我用异丙醇0.3ml/min洗脱C18反向色谱柱后,测试原来的样品结果出现了双峰,压力倒跟洗脱之前没有多大变化,这会是什么问题导致的呢[img]https://ng1.17img.cn/bbsfiles/images/2022/10/202210062045552341_7238_5380090_3.png[/img]
[color=#444444]流动相20%甲醇/水,pH5.9,流速1ml/min。[/color][color=#444444]原先用的安捷伦液相色谱条件基本已成熟,11min出峰且峰形较好。[/color][color=#444444]最近又换了依利特的新机器,3-5min就已出峰,峰形很宽(宽度2-4min)严重拖尾,且出现双峰。[/color][color=#444444]流动相肯定没问题,pH,温度也都一样。[/color][color=#444444]谢谢![/color]







 我要推广仪器
我要推广仪器
 下载APP
下载APP