[color=#444444]现在打算用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定样品中各成分的含量,但由于对照品有些买不到,样品里面成分也很多,不可能买齐所有对照品,买了几个对照品,我想问问用内标法和一两个对照品能不能测出其他成分的含量?[/color]
请问[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]内标法应用有内标物必须与待测物含量相当的要求吗?
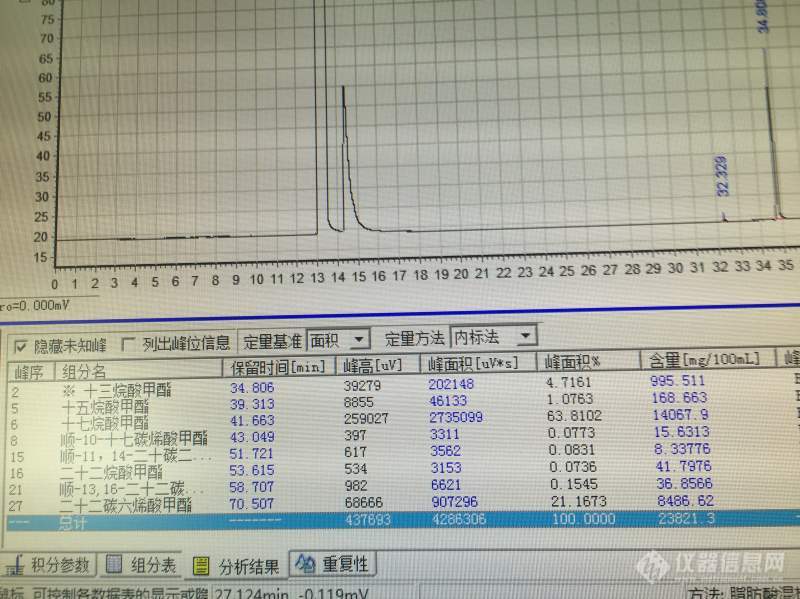
[color=#444444]关于[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]组分含量的问题[/color][color=#444444]目前遇到个关于计算的问题,请各位帮忙解答:[/color][color=#444444]采用十三烷酸为内标,试样加入1ml 的10mg/mL内标物质,跑出来计算如图[/color][color=#444444]我想知道二十二碳六烯酸的含量怎么计算?是用公式C=A/As*Cs吗?[/color][color=#444444]图中的含量(mg/100ml)又是怎么计算出来的?是否还需要自己根据峰面积来计算?[/color][color=#444444][img=,690,516]https://ng1.17img.cn/bbsfiles/images/2019/06/201906171636517194_539_1827556_3.jpg!w690x516.jpg[/img][/color]
[align=center][b][size=18px]运用气相色谱内标法测定食品中甜蜜素含量 [/size][/b] [/align] 甜蜜素化学名称为环己基氨基磺酸钠,于1937年发现,1950年开始生产应用。它是由环己胺和氨基磺酸或三氧化硫反应后用NaOH处理,再重结晶制得的一种白色结晶粉末,甜度为蔗糖的30~80倍。风味较自然,后苦不明显,热稳定性高,是不被人体吸收的低热能甜味剂。1969年曾因其致畸性的报道而被世界各国禁用,后来由于大量试验表明它并无致畸、致癌等作用,许多国家重又许可使用。我国于1987年开始应用甜蜜素,它是目前我国食品行业中应用蕞多的一种甜味剂。甜蜜素含量检测目前有气相色谱检测方法、液相色谱检测方法和液相色谱-质谱/质谱法等,但应用最为广泛的方法是依据GB/T5009.97-2016《食品中环己基氨基磺酸钠的测定》标准中的气相色谱仪分析法。色谱分析技术人员采用在提取溶剂正己烷中加入两种内标物质(甲苯和乙酸正丁酯)对甜蜜素经过衍生后的产物进行定量,具有快捷,准确等特点,受到普遍欢迎。1.试验部分1.1原理在硫酸介质中甜蜜素与亚硝酸钠反应,生成环己醇亚硝酸酯,利用气相色谱法进行定性和定量。1.2试剂(所用试剂不做说明皆为分析纯,水为蒸馏水)1.2.1甜蜜素储备溶液:称取1.0000g甜蜜素(含量≥99.0%),加水溶解并定容至100mL,此溶液浓度为10.00mg/mL,为储备液。置于4℃的冰箱中。本次试验溶液浓度为:10.320mg/mL1.2.2甜蜜素标准使用溶液:取1.2.1储备液10mL,加水定容至100mL,为使用液,浓度为1.0000mg/mL。本次试验使用溶液浓度为:1.0320mg/mL1.2.3100g/L硫酸溶液:称取50g浓硫酸,用水定容至500mL。1.2.450g/L亚硝酸钠溶液:称取25g亚硝酸钠,用水定容至500mL。1.2.5正己烷1.2.6甲苯(内标)1.2.7乙酸丁酯(内标)1.2.8氯化钠1.3仪器1.3.1气相色谱仪GC-2020(带FID检测器、毛细管进样口、N2000色谱工作站)1.3.2DB-5毛细管柱(30m×0.32mm×0.25μm)或其它类型毛细管柱,如DB-1、DB-17011.3.3离心机1.3.410μL微量进样器1.4仪器操作条件1.4.1检测器:200℃1.4.2汽化室:180℃1.4.3柱温:70℃1.4.4载气流速(压力):100kPa1.4.5氢气流速(压力):50kPa1.4.6空气流速(压力):60kPa1.5样品前处理1.5.1内标溶液的配置:在正己烷中加入一定量的内标(乙酸丁酯和甲苯)1,加入的量以出色谱峰合适为宜。一般为500mL正己烷中加入内标100μL~200μL。本次实验乙酸丁酯的浓度为0.1344mg/mL(称量0.0672g乙酸丁酯于装有10mL左右正己烷的25mL容量瓶中,再将溶液转移至500mL容量瓶中并定容至刻度线,摇匀,备用。1.5.2标准溶液前处理:吸取1.2.2标准溶液10mL于50mL比色管中,加10mL水,摇匀,置于冰浴中。加入5mL50g/L亚硝酸钠溶液,5mL100g/L硫酸溶液,在冰浴中放置30min,并时常摇动,然后准确加入10mL1.5.1溶剂,5g氯化钠,摇匀后振摇80次。静置分层。吸出正己烷层于10mL带塞离心管中进行离心分层(如有机溶剂和水相很快就有分层或只要进样针能吸出提取的溶剂,就可以不需离心分离这一步骤),吸取正己烷层1μL注入色谱仪进行分析。算出校正因子2。1.5.3液体样品前处理:称取试样20.0g于50mL带塞比色管中,加10mL水。摇匀,置于冰浴中。处理过程同标准溶液前处理1.5.2。吸取正己烷层试样1μL注入色谱仪进行分析。1.5.4固体样品前处理:称取已磨碎(剪碎)试样2.0~10.0g(根据样品中甜蜜素含量而定称取质量,使甜蜜素的量在1~10mg之间)于50mL带塞比色管中,加10mL水。一些样品不易溶解,如蜜饯类、山楂等,置于水浴锅中煮沸15min左右,冷却至60℃以下,置于冰浴中。处理过程同标准溶液前处理1.5.2。吸取正己烷层试样1μL注入色谱仪进行分析。2结果与讨论2.1.1分析方法的线性相关性的测定分别吸取1.2.1标准溶液1mL、3mL、5mL、10mL、20mL于5个100mL容量瓶中,并定容至刻度,配成浓度分别为0.1032mg/mL、0.3096mg/mL、0.5160mg/mL、1.0320mg/mL、2.0640mg/mL的标准溶液。分别吸取以上标准溶液10mL于5支50mL比色管中,按1.5.2方法进行处理。每个标准点分别进样5次,每次进样1μL,以甜蜜素与内标物的质量比为横坐标,甜蜜素与内标物的峰面积比(取五次进样平均值)为纵坐标绘制标准曲线,得线性方程为y=0.43991x-0.00088,其线性相关系数为0.99999。2.1.2分析方法的准确度的测定从市场上购买一种不含甜蜜素的饮料(样品名称:鲜橙多橙汁饮料生产单位:昆山统一企业食品有限公司净含量:2L生产日期:20060307),称取5份一定量的试样,用移液管分别加入1.2.2标准溶液1、3、5、10、15mL,按上述气相色谱操作条件测定甜蜜素回收率,结果都在99.7%~101.1%之间。2.1.3分析方法的精密度的测定从市场上购买一种含有甜蜜素的饮料(样品名称:鲜橙多生产单位:康裕食品有限公司净含量:1.5L生产日期:20060415),从同一产品中称取五个试样。按1.5.2方法进行处理后,按上述气相色谱操作条件测定甜蜜素含量,得到标准偏差为0.0027,变异系数(RSD)为0.55%。3结论综上所述,本方法简便、快速、准确,具有较高的准确度、精密度和回收率,线性关系好,是一种非常可行的分析方法。注:1用甲苯和乙酸丁酯做内标都可以,可同时加入两种内标(选其中一种作为参照计算),也可以只加其中一种。本次试验的数据是以乙酸丁酯为内标进行计算而获得的。2甜蜜素用本方法衍生处理后,在毛细管柱上会出两个峰,一般是刚开始前面的大,后面的很小,但时间长了,后面的峰越来越大,相应的,前面的峰越来越小直至消失,但两个峰面积和是不变的(48小时之内)。计算校正因子时,用甜蜜素衍生产物的两个峰的面积和计算。检测样品时,两个峰可单独识别并采用同一个校正因子计算,结果相加,也可以使用工作站将峰面积相加,再计算含量。一批样品检测,样品和标准品使用同一浓度的内标溶剂,在计算校正因子时,可将内标物的量假定为1(任一定值皆可),使用非常方便,不需知道加入的内标物的质量。否则,要称量内标物的质量,[font=FZSSK--GBK1-0]精确[/font]至0.0001g,再定容至一定体积。算出内标物浓度,再进行校正因子的计算。
甜蜜素化学名称为环己基氨基磺酸钠,于1937年发现,1950年开始生产应用。它是由环己胺和氯磺酸或氨基磺酸或三氧化硫反应后用NaOH处理,再重结晶制得的一种白色结晶粉末,在高甜度甜味剂中,甜度是最低的,甜度为蔗糖的30~80倍。风味较自然,后苦不明显,热稳定性高,是不被人体吸收的低热能甜味剂。1969年曾因其致畸性的报道而被世界各国禁用,后来由于大量试验表明它并无致畸、致癌等作用,许多国家重又许可使用。我国于1987年开始应用甜蜜素,它是目前我国食品行业中应用最多的一种甜味剂。甜蜜素含量检测目前有气相色谱检测方法和液相色谱检测方法等。最经典的方法是GB/T5009.97-2003《食品中环己基氨基磺酸钠的测定》。但该方法是外标法,且用的是不锈钢填充柱,在没有自动进样器的条件下,要想定量准确,需要花很长时间。要准确地确定一个样品的甜蜜素含量,上机部分至少需要15-~20分钟(要求:进样水平非常高的人),而用内标法只需5~10分钟(要求:一般操作者)。 本文采用在提取溶剂正己烷中加入两种内标物质(甲苯和乙酸正丁酯)对甜蜜素经过衍生后的产物进行定量。 1.试验部分 1.1原理 在硫酸介质中甜蜜素与亚硝酸钠反应,生成环己醇亚硝酸酯,利用气相色谱法进行定性和定量。 1.2试剂(所用试剂不做说明皆为分析纯,水为蒸馏水) 1.2.1甜蜜素储备溶液:称取1.0000g甜蜜素(含量≥99.0%),加水溶解并定容至100mL,此溶液浓度为10.00mg/mL,为储备液。置于4℃的冰箱中。本次试验溶液浓度为:10.320mg/mL 1.2.2甜蜜素标准使用溶液:取1.2.1储备液10mL,加水定容至100mL,为使用液,浓度为1.0000mg/mL。本次试验使用溶液浓度为:1.0320mg/mL 1.2.3100g/L硫酸溶液:称取50g浓硫酸,用水定容至500mL。 1.2.450g/L亚硝酸钠溶液:称取25g亚硝酸钠,用水定容至500mL。 1.2.5正己烷 1.2.6甲苯(内标) 1.2.7乙酸丁酯(内标) 1.2.8氯化钠
[color=#444444]为什么要求内标物含量和待测组分含量一致。我的样品是气体反应,一共有3个产物。其中两个的体积分数占1%左右,另一个占98%。如果我加入了1%的内标物,是不是含量大的物质就会测不准。是为什么?[/color]
有内标物时高效液相色谱法中如何计算含量
高效液相色谱测某样品含量标准品用使用内标进行标定吗高效液相色谱测某样品含量,样品处理后要用内标标记,那做标准曲线用的标准品,在制备时除了用流动相溶解,这同时还用使用内标进行标定吗~~
用GC112A气相色谱测试白酒,内标用乙酸正丁酯,那里有标准的白酒填充柱混标、内标卖。最多组份含量有多少
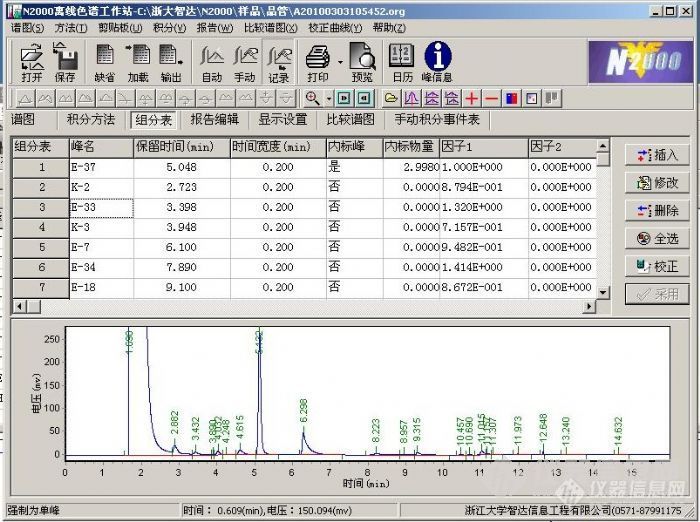
各位学者老师,大家好,小弟又来请教问题了^_^浙大智达的N2000,用内标法进行试验的时候,被测物里面的内标液含量 必须和 标样里的内标物含量 相同。应该是这样吧。除了被测物里面的内标液含量不同以外,其他试验条件完全相同的情况下,如果被 测物的内标液含量 高于 标样里的内标液含量 那做得出的组分含量就下降,反之如果被 测物的内标液含量 低于 标样里的内标液含量 那做得出的组分含量就上升。这么说对吧。我用手动积分修改过被测组分里的内标物峰面积,然后看其他被测组分的含量变动得出的上面的结论。试验是用岛津的色谱做的。请问N2000的软件和岛津不同,请问内标物、组分含量是如何来输入的呢,比如下图内标物是e-37,内标物量为2.9980 单位应该是g,然后其他被测组分是用因子来表示的,然后如何关联标样的谱图呢?逻辑有些混乱见笑啦~标样里的内标物含量应该和被测样品里的内标物含量相等,如果被测物的内标液含量高于标样里的内标液含量(被测组分含量完全一样) ,那做得出的组分含量读数就下降。这样说不知能不能解释清楚。[img]http://ng1.17img.cn/bbsfiles/images/2010/08/201008101708_235762_1919836_3.jpg[/img]
[color=#333333][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法分析汽油中含氧化合物的原始记录,内标物和标定时的含量不同,怎么计算?[/color]
气象色谱 用DMF作内标物,测回收乙醇里的薄荷醇含量!
TCD内标法测甲醇 水 酯中的水的含量 ,现取丙醇作为内标物,配比不同梯度样品浓度的样品做出甲醇-丙醇的校正因子,水-丙醇的校正因子,,发现校正因子不是常数,这个有错误吗,后来想想可能不是线性关系,也许是函数关系,故横坐标取内标物丙醇的质量除以被测组分水的质量,纵坐标取内标物丙醇的峰面积除以被测组分水的峰面积,得出一个函数关系,大部分浓度可以用四次多项式函数拟合处理,相关性为0.986.这种分析方法思路,对吗,恳请各位色谱专家多多建议
用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法自动进样检测溶剂的含量,我准备采用内标法。溶剂为甲醇,异丙醇,乙醇,内标丙酮。如果外标法简单,这几个测得样品混标走个标曲就可以算了。但是内标法怎么测,看到说内标物加入的量应该与待测物一致,但是待测物三个,不知道大致含量。我是否需要先对比丙酮分别先做一个校正因子?那是否需要给丙酮,还有三个溶剂再做个线性呢?
我用内标法(丙酮为内标物)测水中含少量异丙醇,怎样用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]检测异丙醇含量?柱子为PEG-20M,操作步骤及计算方法?
[color=#444444]用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测异丙醇体系中芳香环类化合物(比如苯甲醚,苯乙酮之类)含量,用什么内标物好。求建议,不做这一块知道的很少,请赐教[/color]
在使用液相分析酒内乙酸乙酯和甲醇时,色谱图显示出峰,但是计算含量为零,这是为什么呢?使用的是内标法
用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测定某一物质含量,是否内标法较外标法准确?
[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]采用内标法定性一个杂质,需要内标物与待测杂质的含量一致嘛。比较相对保留时间是样品中内标物与待测杂质保留时间之比与标准溶液中内标物与已知杂质保留时间之一致或在误差范围内嘛(是不是相对保留时间在正负0.1范围内,就能确定待测杂质是我猜测的已知杂质呀)
气质联用-内标法测定新疆石榴籽油中脂肪酸含量摘要:对市售石榴籽油中的脂肪酸化合物成分进行含量测定研究。通过氢氧化钾-甲醇酯化反应,以十一烷酸甲酯为内标物,正己烷为溶剂,采用气相色谱-质谱联用技术对脂肪酸甲酯成分进行分离并辅助NIST检索工具分析成分。建立内标法计算石榴籽油中脂肪酸含量的方法。结果表明,石榴籽油中石榴酸含量超过70% (w/w)。该实验方法简便快捷,可做为内标法测定脂肪酸含量研究提供借鉴。关键词:石榴籽油;脂肪酸;气质联用;内标法中图分类号: 文献标识码:A 文章编号: 石榴(Punica granatum L.)别名安石榴、海榴,为石榴科石榴属落叶灌木或小乔木,在我国亚热带及温带地区均有分布。石榴的果汁、果皮、种籽、花、叶等皆有保健和食疗作用。但是目前对石榴的利用主要在果实和果汁方面。研究表明,石榴籽含油量高达13%-23%,石榴籽油中不仅含有多种不饱和脂肪酸,还含油丰富的甾类、磷脂等成分,具有较好的抗氧化、延缓衰老、降血糖、抗腹泻、预防动脉粥样硬化和减缓癌变进程等作用。随着现代科学技术的进步发展,对石榴籽油的开发和研究已有一定进展,但目前的研究仅集中在面积归一化法测定百分含量上,本文应用气相色谱-质谱联用-内标法对石榴籽油中脂肪酸的种类和含量进行分析测定,本方法简便快捷,色谱分离完全,专属性好,通过本研究可为石榴籽油的脂肪酸含量检测提供可靠的试验依据。1材料与方法1.1 实验材料1.1.1 试验对象 石榴籽油:产地新疆和田地区。1.1.2 主要试剂 标准品:脂肪酸甲酯混合标准品(纯度≥99%);内标物:十一烷酸甘油三酯(5.00mg/mL)。 正己烷、甲醇,由Fisher Scientific公司生产,为色谱纯;氢氧化钾,由天津盛奥化学试剂厂生产,为分析纯。1.1.3 主要仪器 气相色谱—质谱联用仪、配电子轰击离子源,由Perkin Elmer公司生产;分析天平,由OHAUS公司生产;旋涡混合器,由IKA公司生产。1.2 实验方法1.2.1 脂肪酸甲酯化 准确称取酸石榴籽油与甜石榴籽油样品各1.00g 于试管中,分别加入内标溶液2mL(5mg/mL),加入5.0 mL氢氧化钾-甲醇溶液(2 mol/L),40 ℃水浴20 min进行脂肪酸甲酯化,冷却至室温后分别加入5.0mL正己烷进行脂肪酸甲酯萃取,每个样品分别漩涡30s后静置分层后,取上层有机相(正己烷)稀释后用微孔滤膜(0.45μm)过滤后进行气相色谱—质谱仪器进样分析。1.2.2 气相色谱-质谱联用仪条件 色谱柱:FFAP(30 m×0.25 mm×0.5 μm);载气:氦气(99.999%);流速:1.0 mL/min;进样:1.0 μL,分流比1:10;进样口温度:250 ℃;程序升温:初始温度80 ℃,以10 ℃/min升温至230 ℃,保持15 min;离子化方式:电子轰击(EI);离子化能量:70 eV;离子源温度:230 ℃;传输线温度270 ℃;溶剂延迟:2 min;扫描范围:50~450 amu;扫描方式:全离子扫描(SCAN)。1.2.3 石榴籽油中脂肪酸组分定性分析对37种脂肪酸标准进行全离子扫描分析,辅助NIST2011谱图库检索进行脂肪酸甲酯化合物质谱解析并确定各种脂肪酸甲酯的保留时间。通过保留时间及谱图检索匹配脂肪酸的化学结构。1.2.4 脂肪酸组分定量分析对样品的色谱图进行解析,通过内标法进行计算:http://ng1.17img.cn/bbsfiles/images/2015/07/201507221618_556691_1634341_3.bmp; 式中:Xi—样品中脂肪酸甲酯i的含量%;Fi—脂肪酸甲酯i的响应因子;Ai—样品中脂肪酸甲酯i的峰面积;Ac11—样品加入内标物甲酯峰面积;Cc11—内标物浓度(mg/mL);Vc11—样品加入内标物体积(mL);1.0067—十一碳酸甘油三酯转化为甲酯的转换系数;m—样品的质量(mg);Csi—混标中脂肪酸甲酯i的浓度(mg/mL);Ac11—十一碳酸甲酯面积;Asi—脂肪酸甲酯i的峰面积。Cc11—混标中十一烷酸甲酯的浓度(mg/mL)。2 结果与分析2.1 样品前处理方法的确定 目前通过气相色谱法测定脂肪酸需要进行甲酯化反应,常规的甲酯化方法有三氟化硼法、三甲基氢氧化硫法(TMSH)法、重氮甲烷、盐酸-甲醇、硫酸-甲醇和氢氧化铵-甲醇等,以上方法均有一定的局限性。本文采用氢氧化钾-甲醇法进行甲酯化反应,通过多次试验均证明效果较好,该方法操作过程无危害、简单快速、重现性好。2.2 脂肪酸甲酯色谱分离通过对脂肪酸甲酯混合标准进样,优化最佳的色谱条件进行分析得到的色谱图见图1,各组分的保留时间及响应因子Fi值见表1。由此可见混合标准中各种脂肪酸甲酯分离良好,组分之间相互不干扰,已达到定量分析的要求。http://ng1.17img.cn/bbsfiles/images/2015/07/201507221611_556686_1634341_3.bmp图1 脂肪酸甲酯混合标准总离子流色谱图Fig. 1 Fatty acid methyl ester standard of total ion chromatogram 表1 脂肪酸甲酯保留时间及响应因子Table1 Fatty acid methyl ester retention time and response factor序号保留时间化学成分(脂肪酸甲酯)响应因子Fi12.95C6:0已酸甲酯1.42 24.08C8:0辛酸甲酯1.17 36.14C10:0葵酸甲酯1.04 47.24C11:0十一烷酸甲酯1.00 58.33C12:0十二烷酸甲酯0.96 69.41C13:0十三烷酸甲酯0.95 710.45C14:0肉豆蔻酸甲酯0.86 [/t
用内标法进行含量测定,其中标准溶液最近求出的校正值都偏高,是什么原因呢?标准的称样量没问题,标准也 还是之前的标准品,内标溶液也没变,但仪器出来的校正值就比原来一直的数据都高。这样算的含量也有可能偏高的。
我测定元宝枫和雪松的挥发物成分及含量选用3-壬酮,浓度为原液(99%,密度0.82),稀释100倍后,不合适,在稀释液基础上又稀释2倍,这样浓度应该是4.1mg/ml吧。元宝枫进样2ul,出峰较好,相对含量在5%左右;雪松在5ul,出峰较好,相对含量7%。根据内标物计算成分公式:含量mg/g=(组分峰面积/内标物峰面积)*(内标质量mg/样品量g)我理解的内标物质量mg,是进样量*浓度样品量g,是我选择的样品的质量数(叶片1og).如果这样理解的话,内标物的进样量不是很有关系吗?但是我如果用4.1mg/ml浓度在元宝枫中进样5ul的话,内标物占的比例很大35%左右,如果这样计算的话,与2ul的差别是数量级上的。我的上述理解对吗?现在很迷惑。查的资料用的浓度基本在0.41mg/ml的数量上。所以我很迷惑,如果我用4.1的进样2ul合理吗?看了很大资料,发现非化学的专业的,好像很多内标物浓度及计算都不怎么靠谱。如果我认为内标物不准确的话,我直接用峰面积和相对含量对各组分进行比较,应该也是可行的吧?谢谢!
新手,请教各位,采用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]用内标法测定样品中某物质的含量,发现对照品出峰与样品出峰,内标物和所测物质时间都基本一致,只是内标物的峰面积不一致,不知道这样是不是正常?请帮忙!
合成树脂乳液中残余单体含量的气相色谱测定方法探讨 吴亚虎,韩婷婷(广东中山市巴德士化工有限公司品控中心,528427) 摘要:讨论了用气相色谱内标法测定合成树脂乳液中未反应完的残余单体含量的方法,并对该方法的精密度、准确度进行了考察/实验表明,该方法简单易行,准确可靠,适用于各种合成树脂乳液样品。 关键词:气相色谱;极性小口径毛细管柱;内标法; 醋酸乙烯酯、甲基丙烯酸甲酯、苯乙烯、丙烯酸丁酯、丙烯酸异辛酯等单体。 0.引言 自国家十项强制性标准颁布以后,市面上各种涂料中的有害物质必须达到国家相关限量标准。由于合成树脂乳液中未反应的残余单体对人的身体健康和环境会带来不同程度的影响,为此必须设法控制乳液中残余单体的浓度……..目前国科多采用顶空进样技术分析乳液中的残余单体含量,由于仪器投资较大,且样品回收率也不理想,样品前处理较烦锁,对于中小企业这样投资较少,易于在中小企业中推广,该分析方法较难于推广,而我们介绍的是普通分析方法。采用小口径毛细管柱,用氢火焰离子化检测器(FID)进行检测,以内标法定量。柱温采用程序升温,分离效果十分理想。其加标回收率分别在94%-103%之间,分析结果的精密度、准确度完全达到检测要求。 1.实验部分: 1.1 仪器和试剂 电子分析天平(万分之一);GC5890F气相色谱仪(带有分流装置);1.0ul微量进样器;20ml带胶塞小玻璃瓶若干;医用注射器1ml、2ml各两只;小口径毛细柱DB-17HT(0.25mm x 30m x 0.15um;最高使用温度为360℃);积分仪或色谱工作站。醋酸乙烯酯VAM(色谱纯)、甲基丙烯酸甲酯MMA(色谱纯)、苯乙烯ST(色谱纯)、丙烯酸丁酯BA(色谱纯)丙烯异辛酯2-EHA(色谱纯)、丙酮(分析纯)配成4+1混合水溶液(作稀释剂),水(纯净水或蒸馏水)。内标物:环已酮(色谱纯) 1.2 测定原理试样中加适量内标物并用少许丙酮(4+1)稀释摇匀后,用微量注射器将稀释后的溶液注入气相色谱仪,样品被载气带入色谱柱,在柱内被分离成相应的组份,用氢火焰离子化检测器检测并记录色谱图,反数据用内标法计算试样溶液中各待测残余单体的含量。 1.3 测定条件 气化温度:280℃ 检测温度:320℃ 载气:氮气:纯度≥99.99%,变色硅胶+5A分子筛除水、除油,柱前压力为60Kpa(30℃); 氢气:纯度≥99.99%,变色硅胶+5A分子筛除水、除油,柱前压力为65Kpa(30℃); 空气:变色硅胶[/fon
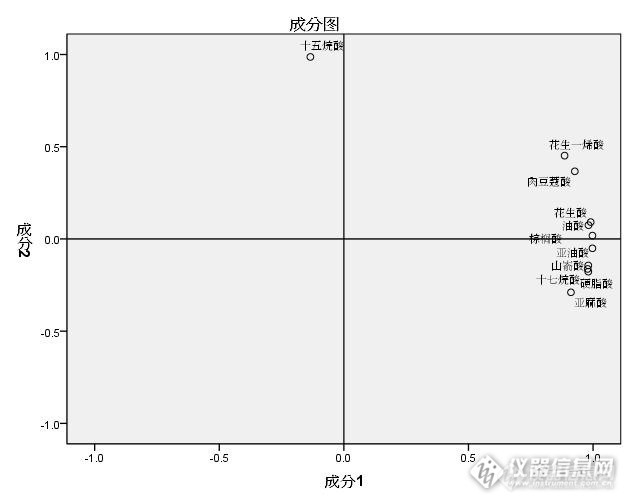
气质联用-内标法测定豆类中脂肪酸含量及因子分析摘要 对市售九种豆类(鹰嘴豆、黄豆、青豆、花芸豆、扁豆、豌豆、绿豆、黑豆、红豆)中的脂肪酸化合物成分进行含量测定研究。通过氢氧化钾-甲醇溶液的酯化反应,以正己烷为溶剂,以十一烷酸甲酯为内标物进行含量计算,采用气相色谱-质谱联用技术对样品中脂肪酸甲酯成分进行分离并辅助NIST检索工具分析成分。通过本次试验建立内标法计算各种豆类中脂肪酸组分含量的方法。结果表明,本次试验的九种豆类样品的脂肪酸组分及含量均有差异,其中青豆的总脂肪含量最高,鹰嘴豆的不饱和脂肪酸含量最高。通过主成分分析获得前2个主成分可以解释全部变异的98%。通过聚类分析将九种豆类归入四大类。该实验方法简便快捷,可为同类产品脂肪酸含量研究提供借鉴。关键词:豆类;脂肪酸;气质联用;内标法;因子分析;聚类分析中图分类号: 文献标识码:A 文章编号: Determination of fatty acids and factor analysis from beans by gas chromatography mass spectrometry using internal standard MethodAbstract: Study on fatty acid composition in nine kinds of beans samples including chickpea、soybean 、 green pea colored kidney bean、dolichos lablab、pea、mung bean、Black bean、Red Bean. Through esterification reaction, using hexane as solvent and quantified by internal standard C11:0 methyl ester. The technology of gas separation of fatty acid methyl ester composition andcomposition analysis of NIST helper tools to retrieve chromatography-mass spectrometry. The results showed, Both fatty acid components and contents are difference between nine kinds of beans samples of this experiment, It can be seen that green beans had the highest total fat content in those samples, chickpeas had the highest content of unsaturated fatty acids. The first principal components obtained by factor analysis could account for 98% of all variations. The single linkage dendrograms based on principal component values divided the nine kinds of beans into four clusters. This test method is simple and fast, which can provide reference for the similar products of fatty acid content.Key words: beans; fatty acids; GC-MS; internal standard method; factor analysis; cluster analysis 当今社会人们对膳食结构的研究日益关注,对食品的要求也从生理需要向营养、食用健康等方面关注,豆类的品种很多,泛指所有能产生豆荚的豆科植物,是中国人的传统食品。各种豆类均含有人体必须的营养成分,有很高的食用和保健价值,可以提高人体的抵抗能力,能有效降低心脏病和癌症的发病几率,目前根据豆类的营养素种类可将它们分为两大类。一类为高蛋白质、高脂肪豆类。另一种豆类则以碳水化合物含量高为特征,本次试验选择九种常见膳食豆类进行脂肪酸含量测定研究,较为系统的对多种豆类进行脂肪酸含量的比较,通过主成分分析和系统聚类分析进行研究,本次测定方法简便快捷,色谱分离完全,专属性好,通过本研究可为豆类样品的脂肪酸含量检测提供可靠的试验依据。1材料与方法1.1 实验材料1.1.1 试验对象豆类:鹰嘴豆、黄豆、青豆、花芸豆、扁豆、豌豆、绿豆、黑豆、红豆,市售。1.1.2 主要试剂 标准品:脂肪酸甲酯混合标准品(纯度≥99%);内标物:十一烷酸甘油三酯(5.00mg/mL)。 正己烷、甲醇,由Fisher Scientific公司生产,为色谱纯;氢氧化钾,由天津盛奥化学试剂厂生产,为分析纯。1.1.3 主要仪器 气相色谱—质谱联用仪、配电子轰击离子源,由Perkin Elmer公司生产;分析天平,由OHAUS公司生产;旋涡混合器,由IKA公司生产。1.2 实验方法1.2.1 脂肪酸甲酯化 准确称取不同豆类样品的均匀粉末2.5g 于试管中,加入内标溶液2mL,加入5.0 mL氢氧化钾-甲醇溶液(2 mol/L),40 ℃水浴20 min进行脂肪酸甲酯化,冷却至室温后分别加入5.0mL正己烷进行脂肪酸甲酯萃取,每个样品分别漩涡30s后静置分层后,取上层有机相(正己烷)稀释后用微孔滤膜(0.45μm)过滤后装进样瓶进行气相色谱—质谱仪器分析。1.2.2 气相色谱-质谱联用仪条件 色谱柱:FFAP(30 m×0.25 mm×0.5 μm);载气:氦气(99.999%);流速:1.0 mL/min;进样:1.0 μL,分流比1:10;进样口温度:250 ℃;程序升温:初始温度80 ℃,以10 ℃/min升温至230 ℃,保持15 min;离子化方式:电子轰击(EI);离子化能量:70 eV;离子源温度:230 ℃;传输线温度270 ℃;溶剂延迟:2 min;扫描范围:50~450 amu;扫描方式:全离子扫描(SCAN)。1.2.3 豆类样品中脂肪酸组分定性分析 对37种脂肪酸标准进行全离子扫描分析,辅助NIST2011谱图库检索进行脂肪酸甲酯化合物质谱解析并确定各种脂肪酸甲酯的保留时间。通过保留时间及谱图检索匹配各种豆类脂肪酸的化学结构。1.2.4 豆类样品中脂肪酸组分定量分析 对测试样品的色谱图进行解析,通过以下内标法公式进行计算: http://ng1.17img.cn/bbsfiles/images/2015/08/201508171137_561066_1634341_3.jpg ;http://ng1.17img.cn/bbsfiles/images/2015/08/201508171137_561067_1634341_3.jpg1.2.5 统计分析方法 本次试验样品均称取3份,每个样品平行测定2次,取平均值进行数据分析,涉及相关统计分析、聚类分析和方差分析方法采用SPSS 22.0软件。2 结果与分析2.1 样品前处理方法的确定 目前脂肪酸的测定试验均需要进行样品的甲酯化反应,常规的甲酯化方法有三氟化硼法、三甲基氢氧化硫法(TMSH)法、重氮甲烷、盐酸-甲醇、硫酸-甲醇和氢氧化铵-甲醇等,以上方法均有一定的局限性。本文采用氢氧化钾-甲醇法进行甲酯化反应,通过多次试验均证明效果较好,该方法操作过程无危害、简单快速、重现性好。2.2 脂肪酸甲酯标准品色谱分析 将脂肪酸甲酯混合标准液进样,通过优化最佳的色谱条件进行分析,由气-质联用仪工作站所得的色谱图见图1,各脂肪酸甲酯组分的保留时间及响应因子Fi值见表1。由标准色谱图可见各组分分离情况良好,内标物与其他组分也相互没有干扰,已达到定性与定量测定分析的要求。http://ng1.17img.cn/bbsfiles/images/2015/08/201508171137_561068_1634341_3.jpg图1 脂肪酸甲酯混合标准总离子流色谱图F[color=#3
请问盐酸多西环素中乙醇含量的检测,采用气象色谱法,按内标法以峰面积比值计算,请问它的具体计算公式是怎样的?
用内标法做定量分析,实际含量是18%,我就只能做到16%。想问问大家有没有关于内标法定量的经验,大家交流一下! 另外,我用的内标物是乙醇,做标准样品时经常经常打不平,希望大家指教!
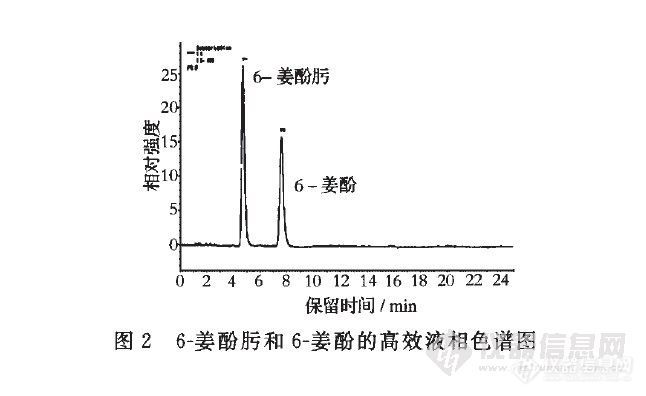
以6-姜酚肟为内标测定生姜及其制品中6-姜酚的含量 曲翔,卢晓旭,黄雪松 (暨南大学食品科学与工程系,广东广州,510632)摘要 建立了一种以6一姜酚肟代替6一姜酚为标样,采用高效液相色谱法,精确测定生姜及其制品中6一姜酚的方法。采用的色谱柱为Diamonsil C18柱,流动相为乙腈一水系统梯度洗脱,流速1mL/min,检测波长280 nm。结果表明:6一姜酚在10.55~2 700 pg/mL、6一姜酚肟在10.43~2 670扯g/mL时峰面积与浓度呈良好的线性关系,相关系数分别为o.998 3和o.999 8。6一姜酚的检测限为2.12 pg/mL,6一姜酚肟的检测限为2.09 pg/mL。6一姜酚对6一姜酚肟的相对校正因子为1.285,相对保留时间为1.728。6一姜酚肟的加标回收率为96.3%~102.2%,相对标准偏差2.4%。用内标法和外标法测得的结果无显著差异,测得鲜姜、干姜和姜酒中6一姜酚含量分别为1.86、3.04、o.115 mg/g。用6一姜酚肟做内标测定6一姜酚的方法准确可靠,可用于生姜及其制品中6一姜酚的测定。关键词 6一姜酚,6一姜酚肟,生姜http://ng1.17img.cn/bbsfiles/images/2012/07/201207242136_379534_2432394_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/07/201207242136_379535_2432394_3.jpg
现车间排放的尾气中含有少量的NMP(N-甲基吡咯烷酮),大概几百PPM,其他都是空气。现领导要求用气相色谱法测试尾气中NMP的含量。现在的难点在于NMP在常温下是高沸点的液体(沸点有200多度),所以用内标法时找不到合适的内标物及气体NMP标样。现在我想到一个方法不知可行不:1.用注射器取定量体积的尾气注射于专门的气体收集袋中;2.再用注射器将与NMP互溶的溶剂(比如丙酮)适量注射于气体收集袋中,充分摇匀,目的是使尾气中的NMP全部溶解于丙酮中;3.再取出溶解有NMP的丙酮,运用常规内标法测试NMP含量。 不知以上方法可行不?我最担心的是丙酮是否能将气体袋中的NMP全部溶解下来?如果不行,那这个方法就是行不通的,请教高手指点,或者用气相色谱法分析此类样品有什么其他更好的办法。谢谢!
用内标法测样品中的含量,内标因子得怎么测,配一个,进三针就行,还是进两针,或者是得配两个或者更多分别进样计算取平均值呢,大家是怎么做的







 我要推广仪器
我要推广仪器
 下载APP
下载APP