推荐几本解质谱比较好的参考书吧
请问二级质谱的碰撞能量是有一个参考范围呢,还是计算出来还是只能自己一点一点摸索?
为了使大家更好地了解质谱的基础知识,光在论坛上进行讨论是不够的。主要还是要静下心来研读相关的书籍。不知道各位在学习质谱的过程中看过的哪写书或文献是值得大家参考的,请大声说出来与大家分享。对于好的建议本版将给予丰厚的积分奖励爱心捐助 爱心捐助 中国心 中国心 [em0818] [em0818]
同种物质,[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]质谱EI模式下的质谱图可以参考液相质谱的电喷雾模式的二级质谱图吗
如题 同种物质,[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]质谱EI模式下的质谱图可以参考液相质谱的电喷雾模式的二级质谱图吗
三重四级杆[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]质谱法测水中农残有什么溯源的标准文件吗?实验室要验证,需要有权威性的参考标准文件才可以,但是我只是找到单四级杆的参考标准,水质相关标准比较滞后而三重四级杆[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]相对是较新技术,所以哪位大侠能提供一些环境相关的有三重四级杆[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用[/color][/url]检测农残的标准文件,谢谢!
我想用液相色谱-质谱联用仪做水环境中污染物的检测,但现在没有标准方法参考,请各位专家介绍几本[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LCMS[/color][/url]应用于水质检测上的专业书籍或者专业期刊,表示感谢!
[font=宋体][url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]分析是建立在参考方法基础上的二次分析方法,因此,以参考方法为基准,从逻辑上[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]方法无法超越参考方法。但模型的预测准确性可以通过增加代表性校正样本数量,采用更合理的计算方法提高。[/font]
所有ASTM方法---使用的各种色谱柱!![em62] [img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/download/search.asp?sel=admin_name&keywords=zjzxwwl2a][b]【分享】《ASTM方法---色谱柱参考简要指南》绝好的资料!!![/b][/url]
故障:调谐时,没有出现参考峰原因(1):参考标样全氟只丁氨瓶中没有参考标样解决方法(1):在质谱仪内置的参考样瓶中添加参考标样全氟砚丁氨原因(2):堵塞了参考标样的管路解决方法(2):将管路拆下并用丙酮超声清洗原因(3):空气泄漏解决方法(3):对空气峰m/z28的高度进行检查,如果比0%氦气峰m/z4的高度大,那么说明有空气泄漏,将丙酮用注射器往各接口处滴加,泄漏的确切位置通过观察丙酮的分子离子峰m/z58的强度变化来进一步的查明
1.故障现象:调谐参数改变时, 调谐峰强度的变化滞后产生故障的可能原因及排除方法:a.离子源被污染,排除方法是对离子源依次用甲醇、丙酮超声清洗各15min;b.预四级杆被污染,排除方法是对预四级杆依次用甲醇、丙酮超声清洗各15min;c.离子源部件未安装到位,电路未接通,排除方法是将离子源拆下,重新安装。2.故障现象:调谐质谱仪时,需要过高的离子能量和推斥电压产生故障的可能原因及排除方法:a.高离子能量过高是由于离子源被污染,推斥电压过高是预四级杆、四级杆被污染,排除方法是对离子源、预四级杆、四级杆依次用甲醇、丙酮超声清洗各15min及保养维护;b. 质谱仪调谐未达到最佳状态,排除方法是重新调谐质谱仪。3.故障现象:调谐参数改变时,仪器响应不明显产生故障的可能原因及排除方法:离子源短路或电路未接通,排除方法是取出离子源, 用万用表测量各部件间的电路连接是否正常。4.故障现象:调谐峰的形状不好,有肩峰产生故障的可能原因及排除方法:a.质谱仪调谐未达到最佳状态,排除方法是重新调谐质谱仪;b.离子源被污染,排除方法是对离子源依次用甲醇、丙酮超声清洗各15min;c.分析器有缺陷或损坏,排除方法是检查分析器外观是否有缺陷或损坏。5.故障现象:调谐时,无参考峰出现产生故障的可能原因及排除方法:a.参考标样全氟只丁氨瓶中无参考标样,排除方法是添加参考标样全氟砚丁氨于质谱仪内置的参考样瓶中;b.参考标样的管路被堵塞,排除方法是拆下管路,用丙酮超声清洗;c.空气泄漏,排除方法是检查空气峰m/z 28的高度,若大于10%氦气峰m/z 4的高度,表明有空气泄漏,用注射器将丙酮滴在各接口处,通过观察丙酮的分子离子峰m/z58的强度变化, 进一步查明泄漏的确切位置。6.故障现象:出现不规则、粗糙的调谐峰产生故障的可能原因及排除方法:a. 离子源被污染,排除方法是对离子源依次用甲醇、丙酮超声清洗各15min;b. 灯丝老化,排除方法是更换灯丝;c.质谱仪调谐未达到最佳状态,排除方法是重新调谐质谱仪。
故障现象:调谐参数改变时, 调谐峰强度的变化滞后产生故障的可能原因及排除方法:a.离子源被污染,排除方法是对离子源依次用甲醇、丙酮超声清洗各15min;b.预四级杆被污染,排除方法是对预四级杆依次用甲醇、丙酮超声清洗各15min;c.离子源部件未安装到位,电路未接通,排除方法是将离子源拆下,重新安装。故障现象:调谐质谱仪时,需要过高的离子能量和推斥电压产生故障的可能原因及排除方法:a.高离子能量过高是由于离子源被污染,推斥电压过高是预四级杆、四级杆被污染,排除方法是对离子源、预四级杆、四级杆依次用甲醇、丙酮超声清洗各15min及保养维护;b. 质谱仪调谐未达到最佳状态,排除方法是重新调谐质谱仪。故障现象:调谐参数改变时,仪器响应不明显产生故障的可能原因及排除方法:离子源短路或电路未接通,排除方法是取出离子源, 用万用表测量各部件间的电路连接是否正常。故障现象:调谐峰的形状不好,有肩峰产生故障的可能原因及排除方法:a.质谱仪调谐未达到最佳状态,排除方法是重新调谐质谱仪;b.离子源被污染,排除方法是对离子源依次用甲醇、丙酮超声清洗各15min;c.分析器有缺陷或损坏,排除方法是检查分析器外观是否有缺陷或损坏。故障现象:调谐时,无参考峰出现产生故障的可能原因及排除方法:a.参考标样全氟只丁氨瓶中无参考标样,排除方法是添加参考标样全氟砚丁氨于质谱仪内置的参考样瓶中;b.参考标样的管路被堵塞,排除方法是拆下管路,用丙酮超声清洗;c.空气泄漏,排除方法是检查空气峰m/z 28的高度,若大于10%氦气峰m/z 4的高度,表明有空气泄漏,用注射器将丙酮滴在各接口处,通过观察丙酮的分子离子峰m/z58的强度变化, 进一步查明泄漏的确切位置。故障现象:出现不规则、粗糙的调谐峰产生故障的可能原因及排除方法:a. 离子源被污染,排除方法是对离子源依次用甲醇、丙酮超声清洗各15min;b. 灯丝老化,排除方法是更换灯丝;c.质谱仪调谐未达到最佳状态,排除方法是重新调谐质谱仪。故障现象:m/z 18、28、32峰大于10%氦气峰m/z 4产生故障的可能原因及排除方法:a. 空气泄漏,排除方法是检漏,检查柱子的连接情况;b. 氦气即将用尽, 气瓶内杂质富集,排除方法是更换载气瓶并安装脱气装置;c. 新近清洗的离子源未烘干,排除方法是设置250℃的离子源温度烘烤离子源;d. 柱子被污染,排除方法是老化柱子。故障现象:灯丝状态良好时,无离子产生产生故障的可能原因及排除方法:a. 离子源需要重新校准,排除方法是利用校准工具重新校准离子源;b. 空气泄漏严重,排除方法是检漏并紧固各连接处。故障现象:调谐质谱仪时, 高质量峰m/z 502、614不显示产生故障的可能原因及排除方法:预四级杆短路,排除方法是将预四级杆拆下, 用氦气或氮气吹干。
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=66777]USP 液相方法参考表[/url]USP 液相方法参考表USP 即美国药典,为高效液相色谱柱填料规定了一些指标,即USP L1---USP L43 系列。用户经常会在文献和技术开发中遇到用符合USP L 系列方法的液相柱分离的要求,为此我们在此把我们公司能够提供的符合此方法的液相柱作一列表,供您参考!
求助大佬,小弟今天做TCEP方法,提取出参考离子,质谱图也可见目标离子,为什么最下面那个框里有红色字提醒(未发现参考离子),也不能积分,我该怎么弄,以前没出现这种情况啊?[img]https://ng1.17img.cn/bbsfiles/images/2020/01/202001112001082896_6698_4050385_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2020/01/202001112001069784_1355_4050385_3.png[/img]
经过数天的努力,我已经亲自测定了某奶粉中的三聚氰胺,参考采用FDA方法见附件,结果检出了三聚氰胺,回收做到77%。采用设备:ABI 3200 串联质谱仪点评:采用苯基柱对三聚氰胺有较好保留,标准曲线配制10-200ppb。检出限约为0.1mg/Kg。还将做进一步方法改进:买的内标——苯代三聚氰胺 刚到,准备下一步改为内标法。欢迎大家跟帖讨论交流。[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=109920]FDAよ猭[/url]
故障现象:调谐参数改变时, 调谐峰强度的变化滞后产生故障的可能原因及排除方法:a.离子源被污染,排除方法是对离子源依次用甲醇、丙酮超声清洗各15min;b.预四级杆被污染,排除方法是对预四级杆依次用甲醇、丙酮超声清洗各15min;c.离子源部件未安装到位,电路未接通,排除方法是将离子源拆下,重新安装。故障现象:调谐质谱仪时,需要过高的离子能量和推斥电压产生故障的可能原因及排除方法:a.高离子能量过高是由于离子源被污染,推斥电压过高是预四级杆、四级杆被污染,排除方法是对离子源、预四级杆、四级杆依次用甲醇、丙酮超声清洗各15min及保养维护;b. 质谱仪调谐未达到最佳状态,排除方法是重新调谐质谱仪。故障现象:调谐参数改变时,仪器响应不明显产生故障的可能原因及排除方法:离子源短路或电路未接通,排除方法是取出离子源, 用万用表测量各部件间的电路连接是否正常。故障现象:调谐峰的形状不好,有肩峰产生故障的可能原因及排除方法:a.质谱仪调谐未达到最佳状态,排除方法是重新调谐质谱仪;b.离子源被污染,排除方法是对离子源依次用甲醇、丙酮超声清洗各15min;c.分析器有缺陷或损坏,排除方法是检查分析器外观是否有缺陷或损坏。故障现象:调谐时,无参考峰出现产生故障的可能原因及排除方法:a.参考标样全氟只丁氨瓶中无参考标样,排除方法是添加参考标样全氟砚丁氨于质谱仪内置的参考样瓶中;b.参考标样的管路被堵塞,排除方法是拆下管路,用丙酮超声清洗;c.空气泄漏,排除方法是检查空气峰m/z 28的高度,若大于10%氦气峰m/z 4的高度,表明有空气泄漏,用注射器将丙酮滴在各接口处,通过观察丙酮的分子离子峰m/z58的强度变化, 进一步查明泄漏的确切位置。故障现象:出现不规则、粗糙的调谐峰产生故障的可能原因及排除方法:a. 离子源被污染,排除方法是对离子源依次用甲醇、丙酮超声清洗各15min;b. 灯丝老化,排除方法是更换灯丝;c.质谱仪调谐未达到最佳状态,排除方法是重新调谐质谱仪。
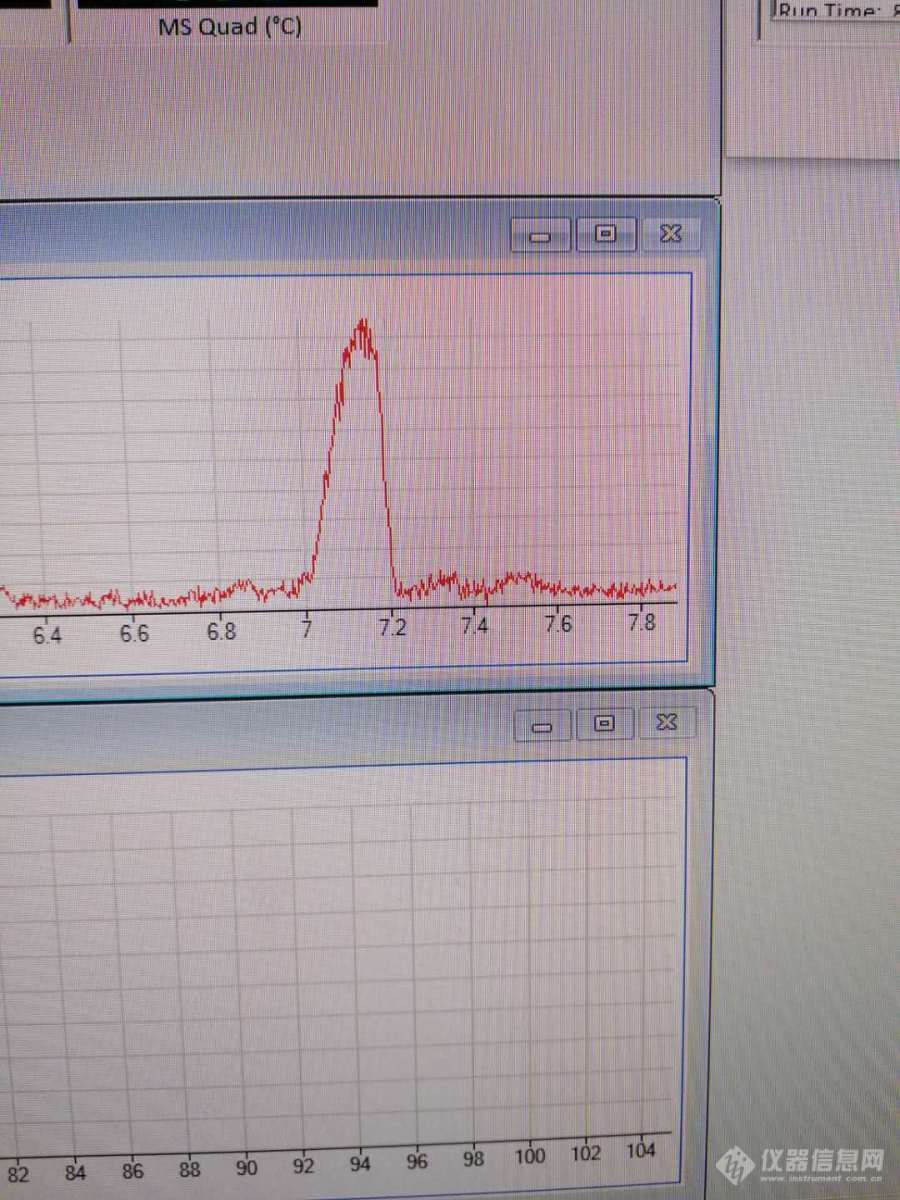
如题,今日在客户现场遇到问题,客户参考GB/T35771-2017开发硫酸二甲酯和硫酸二乙酯[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]方法,标准中使用填料为35%苯基-甲基聚硅氧烷色谱柱,但客户现场没有此种类型的色谱柱,客户使用DB-624代替,两种标准物,20ppm均不出峰。在客户现场做了如下操作:1、检查进样针2、检查进样口,更换进样口分流管线,更换新的衬管,使用甲醇冲洗进样口3、重新配置标样4、质谱调谐正常[b][color=#ff0000]峰形如下:[/color][/b][img=,690,920]https://ng1.17img.cn/bbsfiles/images/2020/09/202009092231387107_6028_3027539_3.jpg!w690x920.jpg[/img]随后,更换HP-5MS色谱柱,峰形响应依然不好求助有没有老师最近做过此项目,使用的色谱柱是否和标准中一致
[color=#444444][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-质谱联用检测某物质残留量,方法学研究需要做哪些内容?除了基本的线性、精密度、准确度(回收率)。[/color][color=#444444]有何参考文件或指导原则?[/color]
化学分析方法验证参考标准,涉及GB 27417、GB 32465、GB 27404,都有关于分析方法验证的内容,但是具体细节和要求不一致?企业实验室,非第三方检验实验室,需要主要按照那个标准执行?另外发布的《食品安全国家标准 化学分析方法验证通则》征求意见稿都快一年了,还未颁布,是否也需要参考这个呢?
做方法验证的精密度结果评价时遇到的问题:我们材料不属于检测标准中给出的参考精密度材料类型。那我的材料有必要小于标准给出的参考精密度才算是证明我们有能力使用该标准方法吗?还是只要给出对于我们自己材料的实验室内精密度就可以?我们是尼龙材料,标准偏差还不大,但相对标准偏差大于标准给的参考值。[img=,690,308]https://ng1.17img.cn/bbsfiles/images/2024/03/202403251628594260_2580_3929349_3.png!w690x308.jpg[/img]
求方法验证和确认的参考。谢谢了
1.故障:调谐时,没有出现参考峰原因(1):参考标样全氟三丁氨瓶中没有参考标样解决方法(1):在质谱仪内置的参考样瓶中添加参考标样全氟砚丁氨原因(2):堵塞了参考标样的管路解决方法(2):将管路拆下并用丙酮超声清洗原因(3):空气泄漏解决方法(3):对空气峰m/z28的高度进行检查,如果比0%氦气峰m/z4的高度大,那么说明有空气泄漏,将丙酮用注射器往各接口处滴加,泄漏的确切位置通过观察丙酮的分子离子峰m/z58的强度变化来进一步的查明。2.故障:m/z1828、32峰比10%气峰m/4大原因(1):空气泄漏解决方法(1):检漏,对柱子的连接情况进行检查原因(2):即将用氦气,杂质富集于气瓶内解决方法(2):对载气瓶进行更换,并对脱气装置进行安装原因(3):新近清洗的离子源没有烘干解决方法(3):设置250摄氏度的离子源温度对离子源烘烤原因(4):污染了柱子解决方法(4):对柱子进行老化3.故障:当调谐参数改变时,调谐峰强度的变化滞后原因:(1)污染了离子源解决方法(1):依次用甲醇、丙酮超声漬洗离子源15分钟。原因(2):污染了预四级杆解决方法(2):依次用甲醇、丙酮超声漬洗预四级杆15分钟。原因(3):离子源部件的安装没有到位,没有接通电路解决方法(3):拆下离子源,重新安装4.故障:调谐参数改变时,仪器响应不明显原因:未接通电路或离子源短路解决方法:将离子源取出,对各部件间的电路用万用表测量,从而判断电路连接是否正常。
[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]分析中的色谱条件,写的是“参考条件”,意思是这个色谱条件可以改动吗?改动幅度多大可以接受呢? 比如我按方法中的条件做出来出峰情况不理想,附近干扰峰影响定量。可以更换色谱柱或者变更升温条件吗?变更后是否还要走什么程序?
论文撰写中利用尾注插入参考文献的方法写论文时,参考文献的引用是一件很麻烦的事,每个杂志要求的文献格式是不一样的,包括在文章中插入的方法和在文章后面排列的格式和顺序等都不同。根据排列顺序,主要分为两种:一是按插入顺序排序,二是按作者的姓名排序。如果是按作者姓名排序,文章内容如果要改动(包括移动、插入或删除),对参考文献在最后的排序影响不大,编号也好改。但如果是按插入顺序排序(国内的绝大部分杂志和国外的许多杂志都是这样的),则文章如有改动,参考文献的增删和重新排序的工作就会变得很烦琐,而且容易出错。有的编辑对这方面的要求很严格,把参考文献的格式作为笔者是否认真的一个重要衡量标准。所以,参考文献是我们写论文时不容忽视的一个环节。 有一个很出名的软件Reference manager是专门用来管理参考文献的,它功能很强大,能对文献进行二次检索、管理,与Word结合还可完成论文中参考文献的插入,相信很多高手都用它解决了参考文献的插入问题。但这个软件不是免费的(D版的不算),而且对于一些不是很高手的人来说,把RM的功能都开发出来也不是件容易的事。我对RM就掌握得不是很好,但我发现其实只要简单地用Word中的插入尾注的功能就能很好地解决按插入顺序排序的论文中参考文献的排序问题。方法如下(以Word2000为例): 1.光标移到要插入参考文献的地方,菜单中“插入”——“脚注和尾注”。 2.对话框中选择“尾注”,编号方式选“自动编号”,所在位置建议选“节的结尾”。 3.如“自动编号”后不是阿拉伯数字,选右下角的“选项”,在编号格式中选中阿拉伯数字。 4.确定后在该处就插入了一个上标“1”,而光标自动跳到文章最后,前面就是一个上标“1”,这就是输入第一个参考文献的地方。 5.将文章最后的上标“1”的格式改成正常(记住是改格式,而不是将它删掉重新输入,否则参考文献以后就是移动的位置,这个序号也不会变),再在它后面输入所插入的参考文献(格式按杂志要求来慢慢输,好像没有什么办法简化)。 6.对着参考文献前面的“1”双击,光标就回到了文章内容中插入参考文献的地方,可以继续写文章了。 7.在下一个要插入参考文献的地方再次按以上方法插入尾注,就会出现一个“2”(Word已经自动为你排序了),继续输入所要插入的参考文献。 8.所有文献都引用完后,你会发现在第一篇参考文献前面一条短横线(页面视图里才能看到),如果参考文献跨页了,在跨页的地方还有一条长横线,这些线无法选中,也无法删除。这是尾注的标志,但一般科技论文格式中都不能有这样的线,所以一定要把它们删除。 9.切换到普通视图,菜单中“视图”——“脚注”,这时最下方出现了尾注的编辑栏。 10.在尾注右边的下拉菜单中选择“尾注分隔符”,这时那条短横线出现了,选中它,删除。 11.再在下拉菜单中选择“尾注延续分隔符”,这是那条长横线出现了,选中它,删除。 12.切换回到页面视图,参考文献插入已经完成了。这时,无论文章如何改动,参考文献都会自动地排好序了。如果删除了,后面的参考文献也会自动消失,绝不出错。 13.参考文献越多,这种方法的优势就体现的越大。在写毕业论文的时候,我就是用这个方法分节插入参考文献的,具爽! 以上就是我用Word中的尾注插入参考文献的方法,拿出来与大家交流一下,请高手们不要见笑。 存在一个小问题: 如果同一个参考文献两处被引用,只能在前一个引用的地方插入尾注,不能同时都插入。这样改动文章后,后插入的参考文献的编号不会自动改动。 解决这个问题其实也不难 1,单击要插入对注释的引用的位置。 2,单击“插入”菜单中的“交叉引用”命令。 3,在“引用类型”框中,单击“脚注”或“尾注”。 4,在“引用哪一个脚注”或“引用哪一个尾注”框中,单击要引用的注释。 5,单击“引用内容”框中的“脚注编号”或“尾注编号”选项。 6,单击“插入”按钮,然后单击“关闭”按钮。 不过得注意:Word 插入的新编号实际上是对原引用标记的交叉引用。如果添加、删除或移动了注释,Word 将在打印文档或选定交叉引用编号后按 F9 键时更新交叉引用编号。如果不容易只选定交叉引用编号,请连同周围的文字一起选定,然后按 F9 键
怎么理解检出限,定量下限,检出浓度和定量浓度,做方法检出限时以哪个为参考最好?[img]https://ng1.17img.cn/bbsfiles/images/2019/02/201902271620493305_3418_3489633_3.png[/img]
XDJM 们,最近我们有个VIP 要和我们要内部方法的参考方法,说是申请CNAS 证书用,我们应该提供么?

[align=center][size=21px]文献方法只做参考[/size][/align][size=16px] 有新的实验项目很多实验员都是先查文献,找检测方法,最后按着自己认为[/size][size=16px]比较靠谱的[/size][size=16px]方法实验。有时[/size][size=16px]也用自己的实验现象、实验结果和文献做个对比,以确定自己实验结果的正确性[/size][size=16px]。[/size][size=16px] 我第一次做饲料中维生素[/size][size=16px]B2[/size][size=16px]检测[/size][size=16px](是在客户实验室帮客户做的)[/size][size=16px]时也查了很多资料,实验方法当时依据的是国标[/size][size=16px]GB/T 14701-2002 [/size][size=16px]饲料中维生素[/size][size=16px]B2[/size][size=16px]的测定[/size][size=16px]。国标中有实验方法,我查资料主要是想看看这个实验有没有需要注意的地方,维生素[/size][size=16px]B2[/size][size=16px]色谱峰保留时间,实验中干扰杂质多不多,该物质响应值高[/size][size=16px]不[/size][size=16px]高等信息。[/size][size=16px]资料查好,准备工作做完后开始实验。[/size][size=16px] 实验时色谱条件参考的国标给的条件[/size][size=16px]色谱柱:[/size][size=16px]2[/size][size=16px]50mm[/size][size=16px]×[/size][size=16px]4.6mm[/size][size=16px]×[/size][size=16px]5[/size][size=16px]μ[/size][size=16px]m[/size][size=16px](当时客户实验室只有这种色谱柱)[/size][size=16px];[/size][size=16px]流速:[/size][size=16px]1.0mL/min[/size][size=16px];[/size][size=16px]柱温[/size][size=16px]:[/size][size=16px]30[/size][size=16px]℃[/size][size=16px];[/size][size=16px]进样量:[/size][size=16px]20[/size][size=16px]μ[/size][size=16px]L[/size][size=16px];[/size][size=16px]检测波长:[/size][size=16px]267nm[/size][size=16px];[/size][size=16px]流动相:在已装入[/size][size=16px]700ml[/size][size=16px]去离子水的[/size][size=16px]1000ml[/size][size=16px]容量瓶中,加入[/size][size=16px]50mgEDTA[/size][size=16px]、[/size][size=16px]1.1g[/size][size=16px]庚烷磺酸[/size][size=16px]钠,待全溶解后,加入[/size][size=16px]25ml[/size][size=16px]冰乙酸、[/size][size=16px]5ml[/size][size=16px]三乙胺,用去离子水[/size][size=16px]定容至刻度[/size][size=16px]摇匀。用冰乙酸、三乙胺调解[/size][size=16px]pH[/size][size=16px]至[/size][size=16px]3.4[/size][size=16px]±[/size][size=16px]0.02[/size][size=16px],过[/size][size=16px]0.45μm[/size][size=16px]滤膜,取该溶液[/size][size=16px]860ml[/size][size=16px]与[/size][size=16px]140ml[/size][size=16px]甲醇混合,超声脱气,待用。[/size][size=16px] 以下是[/size][size=16px]国标色[/size][size=16px]谱条件:[/size][align=left][img]https://ng1.17img.cn/bbsfiles/images/2022/09/202209151928520687_6323_2369266_3.png[/img][/align][align=left][img]https://ng1.17img.cn/bbsfiles/images/2022/09/202209151928525439_1975_2369266_3.png[/img][/align][align=left][size=16px] 按这个方法实验出了一些意外,国标说的保留时间约[/size][size=16px]10min[/size][size=16px],我查的几种文献资料也大概都是[/size][size=16px]8-15min[/size][size=16px],我设的采样时间[/size][size=16px]是[/size][size=16px]15min[/size][size=16px],但[/size][size=16px]15min[/size][size=16px]采样结束,色谱图上没有色谱峰,连续做了几次都没出峰。当时在客户现场,一堆人看着,很是丢人。后来客户下班走了,我自己留在实验室继续琢磨继续做。我首先把色谱柱温度控制[/size][size=16px]到[/size][size=16px]35[/size][size=16px]℃([/size][size=16px]柱温高[/size][size=16px]色谱[/size][size=16px]峰[/size][size=16px]出的[/size][size=16px]快),依然没出峰。于是我又把采样时间改成[/size][size=16px]20min[/size][size=16px],当时怀疑可能是采样时间短没出峰,但[/size][size=16px]依然失望。后又把采样时间改成[/size][size=16px]25min[/size][size=16px]、[/size][size=16px]30min[/size][size=16px],结果依然失望。当时都快晚上[/size][size=16px]12[/size][size=16px]点了,心情很是失落。后我把采样时间设成[/size][size=16px]60min[/size][size=16px],利用这个长时间我重新到网上查找相关资料。找了半天什么也没找到,当时是越找心情越差,那个滋味都不好描述(如果在客户第二[/size][size=16px]天上班前还没解决问题,我都不知道该如何面对客户,毕竟很多[/size][size=16px]文献上都说这个实验难度不大,我也和客户说这个实验没什么难度,保证会做好[/size][size=16px])。正在我失落不知所措[/size][size=16px]的时候[/size][size=16px]奇迹出现了。维生素[/size][size=16px]B2[/size][size=16px]标准品出峰了[/size][size=16px],保留时间[/size][size=16px]31[/size][size=16px]分钟多点,后我又联系做了几次样,保留时间基本一致。[/size][size=16px]后回到公司我换用了多根[/size][size=16px]250mm[/size][size=16px]长,规格和客户现场那根差不多的色谱柱,[/size][size=16px]按照在客户现场实验的色谱条件[/size][size=16px]进行多次实验,发现维生素[/size][size=16px]B2[/size][size=16px]用这个[/size][size=16px]方法,保留时间大概在[/size][size=16px]28-33min[/size][size=16px]之间。[/size][/align][align=left][size=16px] 我不知道国标和那几个文献中的保留时间是怎么做出来的,我做的和他们[/size][size=16px]做的[/size][size=16px]差那么多[/size][size=16px]。[/size][size=16px]我之前一直比较认可各种标准、文献的方法和[/size][size=16px]实验[/size][size=16px]数据等[/size][size=16px],[/size][size=16px]从此发生了[/size][size=16px]改变[/size][size=16px]。[/size][/align][align=left][size=16px] 文献可以参考,但不能完全信赖依赖。[/size][/align]
写论文时,参考文献的引用是一件很麻烦的事,每个杂志要求的文献格式是不一样的,包括在文章中插入的方法和在文章后面排列的格式和顺序等都不同。根据排列顺序,主要分为两种:一是按插入顺序排序,二是按作者的姓名排序。如果是按作者姓名排序,文章内容如果要改动(包括移动、插入或删除),对参考文献在最后的排序影响不大,编号也好改。但如果是按插入顺序排序(国内的绝大部分杂志和国外的许多杂志都是这样的),则文章如有改动,参考文献的增删和重新排序的工作就会变得很烦琐,而且容易出错。有的编辑对这方面的要求很严格,把参考文献的格式作为笔者是否认真的一个重要衡量标准。所以,参考文献是我们写论文时不容忽视的一个环节。 有一个很出名的软件Reference manager是专门用来管理参考文献的,它功能很强大,能对文献进行二次检索、管理,与Word结合还可完成论文中参考文献的插入,相信很多高手都用它解决了参考文献的插入问题。但这个软件不是免费的(D版的不算),而且对于一些不是很高手的人来说,把RM的功能都开发出来也不是件容易的事。我对RM就掌握得不是很好,但我发现其实只要简单地用Word中的插入尾注的功能就能很好地解决按插入顺序排序的论文中参考文献的排序问题。方法如下(以Word2000为例): 1.光标移到要插入参考文献的地方,菜单中“插入”——“脚注和尾注”。 2.对话框中选择“尾注”,编号方式选“自动编号”,所在位置建议选“节的结尾”。 3.如“自动编号”后不是阿拉伯数字,选右下角的“选项”,在编号格式中选中阿拉伯数字。 4.确定后在该处就插入了一个上标“1”,而光标自动跳到文章最后,前面就是一个上标“1”,这就是输入第一个参考文献的地方。 5.将文章最后的上标“1”的格式改成正常(记住是改格式,而不是将它删掉重新输入,否则参考文献以后就是移动的位置,这个序号也不会变),再在它后面输入所插入的参考文献(格式按杂志要求来慢慢输,好像没有什么办法简化)。 6.对着参考文献前面的“1”双击,光标就回到了文章内容中插入参考文献的地方,可以继续写文章了。 7.在下一个要插入参考文献的地方再次按以上方法插入尾注,就会出现一个“2”(Word已经自动为你排序了),继续输入所要插入的参考文献。 8.所有文献都引用完后,你会发现在第一篇参考文献前面一条短横线(页面视图里才能看到),如果参考文献跨页了,在跨页的地方还有一条长横线,这些线无法选中,也无法删除。这是尾注的标志,但一般科技论文格式中都不能有这样的线,所以一定要把它们删除。 9.切换到普通视图,菜单中“视图”——“脚注”,这时最下方出现了尾注的编辑栏。 10.在尾注右边的下拉菜单中选择“尾注分隔符”,这时那条短横线出现了,选中它,删除。 11.再在下拉菜单中选择“尾注延续分隔符”,这是那条长横线出现了,选中它,删除。 12.切换回到页面视图,参考文献插入已经完成了。这时,无论文章如何改动,参考文献都会自动地排好序了。如果删除了,后面的参考文献也会自动消失,绝不出错。 13.参考文献越多,这种方法的优势就体现的越大。在写毕业论文的时候,我就是用这个方法分节插入参考文献的,具爽! 以上就是我用Word中的尾注插入参考文献的方法,拿出来与大家交流一下,请高手们不要见笑。 存在一个小问题: 如果同一个参考文献两处被引用,只能在前一个引用的地方插入尾注,不能同时都插入。这样改动文章后,后插入的参考文献的编号不会自动改动。 解决这个问题其实也不难 1,单击要插入对注释的引用的位置。 2,单击“插入”菜单中的“交叉引用”命令。 3,在“引用类型”框中,单击“脚注”或“尾注”。 4,在“引用哪一个脚注”或“引用哪一个尾注”框中,单击要引用的注释。 5,单击“引用内容”框中的“脚注编号”或“尾注编号”选项。 6,单击“插入”按钮,然后单击“关闭”按钮。 不过得注意:Word 插入的新编号实际上是对原引用标记的交叉引用。如果添加、删除或移动了注释,Word 将在打印文档或选定交叉引用编号后按 F9 键时更新交叉引用编号。如果不容易只选定交叉引用编号,请连同周围的文字一起选定,然后按 F9 键。
国家标准中规定的气相色谱参考条件可以作修改吗?
[size=4]endnote默认的是,在每一个word文档的末尾生成参考文献的目录,那如何在每一章节后面生成参考文献的目录,并且每一章节重新编号呢?这个问题困扰了好多人好长时间……解决的方法有两种:第一种解决办法:这是一种最简单的办法,适合于任何版本的endnote软件。具体操作如下:(1)每一章节单独作为一个word文档进行写作,键入参考文献(2)定稿后,去除域代码,生成终版的单独的word文档(3)将单独的word文档合并即可实现最初的愿望备注:该方法的特点是:生成word文档偏多,但是计算机处理速度较快。[/size]







 我要推广仪器
我要推广仪器
 下载APP
下载APP