请问大家在用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]建立分离分析方法时,都优化哪些条件,从哪些方面考虑的呢?多谢!
请问色谱测量甲苯的方法和条件是什么?
我做有关物质的结构鉴定,优化好色谱条件后,质谱上却没有响应,由于刚刚开始做MS,想请教各位提高离子响应的方法有那些啊?
[center]液相色谱测VFA的条件及方法[/center] 本人在做厌氧发酵实验,实验过程中产生的VFA是本实验的重点,因此需测其VFA组分,现在恳请各位高手帮忙教我如何通过液相色谱测其VFA组分,条件及步骤等,十分感谢!
我用的是岛津的[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]质谱联用,想要做尽可能多的TCA循环,糖酵解相关的小分子有机酸,只需要知道相对值,这样的话只能做非靶向的scan模式吗?我之前用的scan条件在质谱库自动检索基本检测不到想要的物质,而且峰形也不太好,但是不知道如何改进方法。网上也搜索不到前处理和目标物质都相似的文章,请问样品处理时的溶剂和方法也会影响色谱条件吗?不同品牌仪器的方法参考意义大吗?感谢各位老师同行赐教
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]检测枸杞中乐果、对硫磷、马拉硫磷的方法,色谱条件,能给个图最好
液相色谱分析间苯二磺酸和苯磺酸的条件及定量方法?
液相色谱测定水中甲酸乙酸方法条件是什么?
不知谁能分享检测饲料中恩诺沙星色谱条件及检测方法。
请高手指点PBBs、PBDEs的液相色谱条件及方法,主要是测定塑胶中的多溴联苯和多溴联苯醚,不知道样品的前处理方法及色谱条件如何。望高手指点,小弟万分感谢!
1,3-丙二醇 液相色谱测定方法条件,请问有那位大侠做过,急用,谢谢!
做扩项的时候验证报告里会有个色谱条件。后来发现还能优化,认证时候做盲样或者加标时候能用已经优化的色谱条件吧,还是必须用方法验证报告里的色谱条件?

液相色谱-串联质谱条件测定水果中的多农药残留方法优化本法建立了水果多种农药残留量的快速、简便、准确的测定方法,通过对样品前处理方法、仪器检测方法的考察优化,建立液相色谱-串联质谱检测方法。其中以日本制定的“肯定列表”中的“一律标准”最为严格,限量为 0.01mg/kg,而我国的残留限量标准还不够完善,很多农药还没有制定限量标准,其方法的测定低限(定量限)能达到 0.01mg/kg的要求。1、液相色谱条件考察:在方法建立过程中对液相色谱条件进行了考察,主要考察了色谱柱、流动相等。在色谱柱选择时,比较了BEH C18、 HSS T3、 Zorbax Eclipse Plus C18等色谱柱,发现HSS T3色谱柱对甲胺磷、乙酰甲胺磷等大极性农药的色谱保留效果较好,所以选择HSS T3色谱柱进行下一步的研究。在流动相考察时,发现在流动相中加入0.1%的甲酸可以改善多菌灵、噻菌灵等农药的分离,而且加入甲酸可以在电喷雾正离子( ESI+)模式电离时提供H+,提高电离效果,所以选择在流动相中加入0.1%的甲酸,比较了在水相和有机相中均加入0.1%的甲酸、仅在水相中加入0.1%的甲酸两种情况,发现色谱分离及质谱电离无显著差别,为简化操作和便于使用,选择仅在水相中加入0.1%的甲酸。流动相的有机相选择时考察了甲醇、甲醇-乙腈( 1+1, V/V)、乙腈三种情况,发现采用乙腈时色谱柱的柱压较低,色谱分离也较好,但毒死蜱、辛硫磷、特丁硫磷、敌敌畏等常用有机磷农药的质谱响应值低、重现性差,而选用甲醇时可以显著提高这些化合物的质谱响应及重现性,综合考虑后选择甲醇为流动相的有机相。由于此次分析的多农药的化学性质差别较大,从高极性到低极性均有分布,所以色谱分离时需要采用梯度洗脱模式,通过实验考察,最终确的液相色谱条件如下:a)色谱柱: HSS T3柱,长100 mm,内径2.1 mm,粒径1.8 μm,或相当者; b) 流动相:甲醇-0.1%甲酸溶液梯度洗脱,参见表 1。 https://ng1.17img.cn/bbsfiles/images/2020/09/202009211516176216_1219_2166779_3.png!w607x264.jpgc) 柱温: 35 ºC
对于单甘酯类的物质用什么色谱条件分离比较好呢?老师给我的方法用的是正己烷:异丙醇=10:1,流速0.7ml/min,正相色谱柱分离。示差折光检测器。不知道怎么样。同样的色谱条件用蒸发光散射检测器怎么样?望高人指点一下。
各位大侠:谁知道高纯氢和高纯氮的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析方法,色谱仪器配置,色谱分析条件,谢谢!
菜鸟求教:用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析同一种物质时,如果样品制备方法不同,那么色谱条件是不是也就要不一样呢?
老师们好,最近在进行指纹图谱的相关实验,产生了一个疑问,希望老师们能帮忙解答一下。在实验中,应该先优化样品前处理的条件还是色谱分析的条件呢?若先优化前处理条件,该用什么色谱分析方法对处理好的样品进行分析呢?若采用的色谱分析方法和最后优化的色谱方法不同,会不会导致优化时变量不唯一的情况?
如题。有时候,因为分离的不理想,会对规定的色谱条件作一些小的修改,这时候要做方法验证吗?
[table=100%][tr][td]求助:采用超高效液相色谱法进行一种药物的方法学考察,经过尝试,在一种色谱条件下可以得到药物的色谱峰,但峰面积较小,与浓度不符,此时应该更改哪个色谱条件?还是跟标准品溶解时的溶剂有关?[/td][/tr][/table]
菜鸟求教:用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析同一种物质时,如果样品制备方法不同,那么色谱条件是不是也就要不一样呢?
由于这几天都在做用SCX色谱柱进行样品检测,有个问题想要和大家讨论一下,在使用离子交换色谱法中,怎么优化色谱条件?离子交换色谱柱法:是以离子交换剂为固定相如:强阳离子交换柱(SCX),强阴离子交换柱(SAX),以缓冲溶液为流动相,借助于试样中电离组分对离子交换剂亲和力不用以达到分离离子型或可离子化的化合物的目的。我就抛砖引玉了:1、改变缓冲盐的浓度,一般增加缓冲盐的浓度可以缩短保留时间;2、改变pH值,一般在碱性条件下强阳离子交换柱(SCX)色谱法中,保留时间会缩短;3、……大家来说说自己的观点吧
测果汁中的有机酸,苹果酸,柠檬酸,莽草酸,奎宁酸,请问合适的条件和方法是什么?比如说用什么样的柱子?用液相还是离子色谱?流动相是什么等。谢谢大家!

药典方法红花含量测定使用两种方法分别测定羟基红花黄色素A和山奈素两种成分含量色谱条件如下:[img=,477,96]https://ng1.17img.cn/bbsfiles/images/2020/12/202012101605485344_5670_2204446_3.png!w477x96.jpg[/img]如果用一种流动相同时测定两个组分,可分别采集403nm,367nm两个通道信号。色谱条件如何优化?
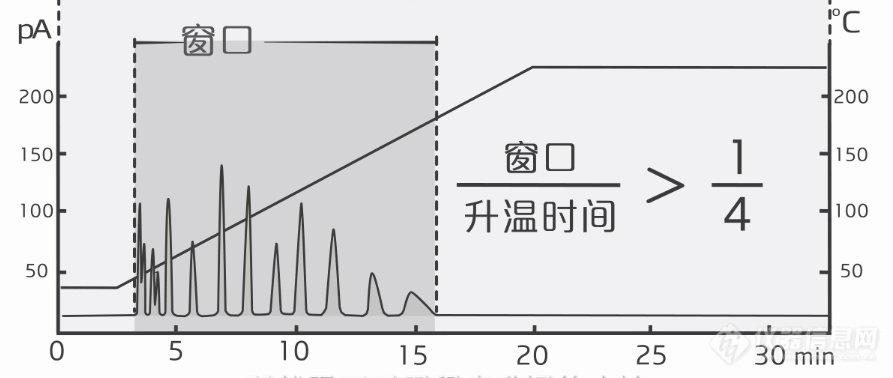
[align=center][/align]在[b][back=yellow]恒温方法[/back][/b]中,色谱柱的温度保持不变。它对于沸点差异不大,保留性质差不多的化合物组比较有效。但是,当要分析的物质保留性质差异较大时。较晚出的色谱峰就会出现峰展宽、峰型变差、灵敏度降低等各种问题。而较早出的色谱峰又很难达到满意的分离度。这时候我们就需要用程序升温方法了。在[b][back=yellow]程序升温[/back][/b]的方法中,柱温起始温度较低,让较早出的色谱峰实现比较好的分离。接下来温度上升,并在较高温度下保持一段时间,可以让后出的色谱峰整体前移,缩短分析时间。这样保留较强的化合物出峰提前,实现更窄的峰宽和更高的灵敏度。[color=red]一般采用下面这个标准的测试升温梯度,作为条件优化的起始:初始温度尽量低,比实验室室温高15度就可以了。然后,以每分钟10度的速度升温。最后在色谱柱最高耐受温度以下20度左右停留10分钟,观察所有进样的化合物都能够以不错的分离度全部跑出色谱柱。然后根据得到的色谱图对条件进行进一步的优化。[/color]如果所有峰出峰时间的“窗口”小于程序升温时间1/4,那这个方法就可能考虑采用恒温方法。建议恒温温度比最后一个峰的出峰温度低45度。接下来可以以10度左右上下调整,以得到最满意的分离度。如果所有峰出峰时间的“窗口”大于程序升温时间1/4,或者恒温分析无法获得满意的分离度,那就需要采用[b][color=#ff6666]程序升温[/color][/b]的方法。[align=center][img=,690,292]https://ng1.17img.cn/bbsfiles/images/2022/03/202203041911518047_6826_4079281_3.png!w690x292.jpg[/img][/align]对于[b]不分流进样[/b]来说,初始柱温通常设定的比溶剂沸点低10到20度,保持1分钟。因为在不分流模式下,衬管中的气体流速很小,样品需要较长的时间才能跑到色谱柱里,因此,需要使用较低的初始柱温,利用溶剂聚焦来获取更窄的峰宽。对于[b]分流进样[/b],衬管里的载气流速很快,样品几秒钟内就会从衬管转移到色谱柱。所以从样品聚焦的角度,对初始温度没有特别的要求。有时候可以稍微降低初始温度和延长保持时间,来改善靠前出的峰的分离度。其次,是[back=yellow]升温速率[/back]的设定,它对靠中间出的峰的分离度有巨大的影响。一般来说,升温速率越快,出峰越快,分析时间越短,但是会降低峰的分离度。所以这是一个效率和效果的博弈,我们期望用最短的分析时间,达到满意的分离度。一般来说,推荐的升温速率可以用这个公式来计算:升温速率=10/T[sub]0[/sub],这里的T[sub]0[/sub]为死时间,就是不保留的化合物在色谱柱上的出峰时间。接着是恒温平台的设定。如果用同一个升温速率,中间有些峰仍然无法实现很好分离的话,可以在这些峰出峰温度以下45度左右,设置一个恒温平台,保持2到5分钟后继续升温,这样能够实现更好的分离。对于复杂的样品,可能需要设置多个升温的平台,才能实现最优化的分离。[align=center][img=,690,317]https://ng1.17img.cn/bbsfiles/images/2022/03/202203041915120859_4895_4079281_3.png!w690x317.jpg[/img][/align]最后是结束温度的设定。升温最高的温度一般设置为最后一个色谱峰出峰温度以上20度左右。但是对于有些复杂基质的化合物分析,我们会把程序升温的结束温度设定的更高一些,对色谱柱进行一段时间的烘烤,防止有些高沸点的化合物在色谱柱中残留,影响后续的检测。但是要注意不能超过色谱柱的最高耐受温度。
硝基苯色谱操作条件柱温:180℃ 汽化、检测温度:240℃量程:2 载气总压:300Kpa 载气分压:70 KPa 氢气压力:70 KPa空气压力:70 KPa 进样量:0.5μl 处理机衰减:2 纸速:3方法:45/44 进样器:2#文件号(成品):2#文件号(中控):0# 焦油:4#苯胺色谱操作条件检测器:氢火焰离子化检测器进样量:1μl 文件号:2衰减: 纸速:柱起始温度:125℃ 柱初温时间:12 min 升温速率:10℃ /min柱终止温度:200℃ 柱终温保持时间:5 min 汽化温度:250℃检测温度:250℃ 载气压力:160Kpa 空气压力:50 Kpa氢气压力:60 Kpa 分流气流量:27ml/min隔垫吹扫气流量:4 ml/min硝基苯色谱操作条件检测器:氢火焰离子化检测器进样量:1μl 文件号:7 衰减:2 纸速:3柱起始温度:80℃ 柱初温时间:0min 升温速率:8℃ /min柱终止温度:180℃ 柱终温保持时间:10 min 汽化温度:200℃检测温度:300℃ 载气压力:140Kpa 空气压力:50 Kpa氢气压力:60 Kpa 硝基氯苯色谱操作条件柱温:135℃ 汽化室温度:240℃ 检测器温度:230℃ 载气总压:380kPa 载气1#分压50 kPa 2#分压50 kPa量程:101 1#柱:邻硝 文件号:2 衰减:4 纸速:52#柱:对硝 文件号:1 衰减:4 纸速:5氯苯色谱操作条件柱温:120℃ 汽化室温度:180℃ 检测器温度:180℃ 载气总压:140kPa 载气1#分压50 kPa (水份) 2#分压40 kPa(氯苯分析) 桥流:140mA 文件号:0#(氯化液)1#(成品)3#(焦油)衰减:2 纸速:5 方法号:45硝基氯苯色谱操作条件柱温:135℃ 汽化温度:240℃ 检测温度:230℃ 载气总压:300kpa分压:50 kpa 氢气:60 kpa空气:50 kpa 检测器:1#(FID)进样器:1# 衰减:2纸速:3文件号:1#(对硝) 2#(邻硝) 3#(中性一硝)4#(邻对位低油氯苯废水及中性一硝色谱操作条件柱温:120℃ 汽化温度:210℃ 检测温度:210℃ 载气总压:300kpa空气:50 kpa 氢气:50 kpa衰减:3 纸速:3 量程:102 方法号:45废水 :进样器:1# 进样量:0.5ul 文件号:6# 分压:40 kpa 极性:+苯:进样器:1# 进样量:0.2ul 文件号:4#中性一硝及塔釜进样器:2# 进样量:0.5ul 文件号:2#、3# 分压:150kpa 极性:-苯胺色谱操作条件柱温:130℃ 汽化温度:210℃ 检测温度:210℃电流:0.5nA 载气分压:50kpa 量程:101处理机衰减:3 进样量:0.2ul方法号:44 纸速:1 文件号:1#氯化苯(苯胺)水份色谱操作条件柱温:120℃ 汽化温度:180℃ 检测温度:180℃桥流:140mA 载气总压:120kpa载气分压:35kpa进样器:氯化苯1#;水份:2# 处理机衰减:2进样量:水份1ul,氯化苯含量2 ul方法号:45纸速:5 文件号:成品(0#);氯化液(1#)附温度、浓度对照表温度℃6789101112131415饱和水%0.03380.03530.03680.03840.040.04170.04350.04520.04720.0491温度℃16171819202122232425饱和水%0.05130.05350.05560.05810.06070.06320.06580.06830.07130.0742温度℃26272829303132333435饱和水%0.07740.08070.08390.08780.09160.09550.09930.10320.10760.112氯化苯(苯胺)水份色谱操作条件柱温:120℃ 汽化温度:180℃ 检测温度:180℃桥流:140mA 载气总压:120kpa载气分压:35kpa进样器:氯化苯1#;水份:2# 处理机衰减:2进样量:水份1ul,氯化苯含量2 ul方法号:45纸速:5 文件号:成品(0#);氯化液(1#)附温度、浓度对照表温度℃6789101112131415饱和水%0.03380.03530.03680.03840.040.04170.04350.04520.04720.0491温度℃16171819202122232425饱和水%0.05130.05350.05560.05810.06070.06320.06580.06830.07130.0742温度℃26272829303132333435饱和水%0.07740.08070.08390.08780.09160.09550.09930.10320.10760.112氢气色谱操作条件柱温:40℃ 汽化温度:50℃ 检测温度:50℃桥流:100mA 载气总压:100kpa载气分压:40kpa进样器:1# 处理机衰减:0进样量:2ml方法号:44纸速:3H2﹦100-O2-N2
我想测一种厌氧氨氧化产生的气体中氮气的含量,大家能否指点一下,怎么用气相色谱测氮气的含量。用TCD检测器 TDX01的色谱柱测试的各个条件是什么?用什么做载气?标准气体是什么?
总是有人看到某个贴子的做个样品,就会问你们的仪器条件是怎么样设置的,用什么柱子,我用的条件与柱子和你的一样但是我们峰出的就没有你的好呢?这是为什么呢?其实做色谱分析,我觉得没有一个条件是固定的,不管你用的是同一台仪器,同一个柱子,都有可能因为样品的特性导致峰型不好,那就要根据不同的样品重新设定不同的条件,同是为什么会有人说我们都是做标液为什么他们的峰型都很好我们的就不好,那么大家有没有考滤过你的仪器状态与前者是否一样?你的柱子长度是否与前者一样?你的柱效是否也与前者一样呢?所以说每个方法都不是一个固定的,都是只能根据不同的条件随时发生变化的,文献,标准的方法条件都只能做为参考http://simg.instrument.com.cn/bbs/images/default/emyc1002.gif
大家好,我是一名[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]新手,在建立一个色谱条件下要做一个方法学验证,方法学验证过程中需要做加样回收率试验,我们现在规定加样试验要连续进18针加样溶液,但依据我目前的色谱条件(柱温最高200度保持10min),在进完5、6针过后会出现样品的残留,接下来谱图的基线就会受到影响,直接影响我所要的溶剂峰面积。我想问在这种情况下,是提高柱温还是增加最高柱温保留时间,亦或是两者都增加。
柱子:40米毛细柱,0.32mm,膜厚0.33um柱前压:15/25psi,分流口:10/67ml/min柱温:50℃保持2min,10℃/min升到100℃,检测器温度250℃,进样器温度250℃;问题:用1000ul/ml的苯系物标液(包括溶剂甲醇,苯、甲苯、二甲苯、苯乙烯、乙苯)进样0.2ul、0.4ul、0.6ul、0.8ul、1.0ul,得到标准曲线,分析100ul/ml的标液(盲样)结果却是160ul/ml左右。(两台设备两人结果都如此)求助:问题出在哪?有没有好的分析条件?(前提是要求色谱图在10分钟以内/最好7、8分钟出完)加急!!!!
请问用液相色普法检测面粉中过氧化苯甲酰(增白剂)的前处理方法及色谱条件。急切盼回!!!







 我要推广仪器
我要推广仪器
 下载APP
下载APP