现在需要对氨气、氢气和氮气混合气中各个气体的含量进行检测,请问[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]质谱中该使用什么规格的分离柱、检测器和分析柱,还有载气用什么好?
我所工作的乳业单位想要开展气相色谱法检测氮气,不知哪位高手可以指点一二?检测方法,所需设备型号,前处理注意事项,检样批次等等,都可以知无不言,言无不尽,晚辈先谢过各位大侠义气相助!!!
请色谱专家们推荐一下,检测氩气中氮气和氧气含量的气相色谱型号和检测器。谢谢
我公司有部门想检测氨气中所含有的氧气与氮气的含量,其中可以知道的是氨气含量已经达到了90%上以,是否可以用色谱进行检测?还有氨气对色谱柱是否有影响?
安捷伦的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url](TCD检测器),载气为:氢气,支持气:氮气。为什么需要支持气啊?FID检测器中的氮气是载气,氢气与空气是氢火焰燃烧要用的。我就弄不明白TCD检测器为什么需要氮气做支持气。
图便宜买了一个说是进口的,后来买来才发现是国内组装的氮气发生器,也没有相关的进口文件和标志什么的,放质谱上用不了,没有诚信。只能放氮吹用。唉~有没有推荐的进口品牌,不想再用膜的了!
不知[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]能不能检测氮气的含量,在几十到几百ppm量级吧,如果不能,那有没有什么仪器可以检测?
生物发酵涉及到医药、轻工、食品、农业、海洋、环保等众多领域,在我国国民经济发展中占有极其重要地位,是当前经济社会发展急需突破的技术领域,也是当前世界各国发展的热点领域。在生物发酵过程中,对发酵尾气中各种气体组分的检测有着相当重要的地位。发酵尾气的组分变化,反映了整个发酵过程中物质的变化情况,对尾气数据的分析,可对发酵过程起到监测的作用。 在项目完成过程中,项目组根据发酵尾气的特点以及现场应用环境的要求,对尾气预处理、采集、分析、数据处理等进行了一系列的条件优化,最终建立了一套“在线质谱仪直接分析生物发酵尾气的方法”和标准操作程序。采用SHP8400PMS在线质谱仪可对发酵尾气进行直接分析,实现实时自动在线监测,能够获得连续稳定的准确测量结果,对氧气、二氧化碳、氮气、氩气以及各种挥发性的物质进行高精度定量分析,提高了监测效率。目前该方法已成功应用于国家生化工程技术研究中心(上海)的发酵工程研究和多家生物制药企业的生产现场监测,具有推广应用的示范意义,为建立行业标准方法打下基础。专家组在给予项目肯定和高度评价的同时,也提出了相当中肯的进一步研究建议,希望能将国产质谱仪更好的应用于现场监测领域。
我们实验室用氮气发生器提供氮气。但是头几天发生器突然不工作了,好在实验室有人,把质谱仪关了。想问如果这样的事发生在晚上或者周末,对质谱会有什么样的损害?氮气同质谱的真空系统有什么关系吗?谢谢。。。。。感谢大家的回答。我用的是Bruker 的 Q-TOF. 有机会我再向工程师确认一下。要是有更确定的信息我再贴上来~~
[color=#444444]最近在做一个项目,得到了一个产品的二氯甲烷溶液,分别用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用[/color][/url]和直接测的质谱检测了分子量,但是两个检测结果差别很大。[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用[/color][/url]的检测结果中有我目标产物的分子量,但是直接测的质谱里根本看不到我目标产物的分子量,局部放大了看,也没有啊。[/color][color=#444444]我一直以为这两种质谱检测的结果应该一致才对啊,所以现在我很迷茫,不知道这是什么原因,求高人指教![/color]
[size=4][color=#DC143C][font=黑体]同位素比质谱方法检测内源性类固醇雄烯二酮[/font][/color][/size]=========================================在所使用的禁用物质中,类固醇激素是较为普遍使用的一类药物。人体自身能合成与分泌的类固醇激素称为内源性类固醇激素,如攀酮。由于在检测方法上有一定难度,一些运动员选择使用内源性类固醇制剂以逃避兴奋剂检测。目前,兴奋剂检测实验室应用同位素比质谱分析方法检测内源性类固醇来源。13C和12C是碳元素在自然界中的天然同位素。有机化合物的来源不同,其同位素比(如13C与12C的比值)也不同。人体自身分泌的类固醇与相同化学结构的类固醇制剂的同位素比不同。应用同位素比质谱分析技术可以测定化合物13C与12C的比值,同位素比用δ(‰)值表示。根据仪器的分析流程和组成部分,本文方法称为[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]/燃烧炉/同位素比质谱([url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/C/IRMS)方法。该方法在兴奋剂检测中的应用时间较短,文献方法较为繁琐,本文建立了快速灵敏的[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/C/IRMS分析方法。-------------------------------------------------试验材料与方法1. 试剂和对照品试剂:β-葡萄糖醛酸酶,Sigma;其余均为国产分析纯试剂。对照品:睾酮(缩写T)、雄酮(缩写An)、本胆烷醇酮(缩写Etio)、5α-雄烷-3α,17β-二醇(缩写5α-diol)、5β-雄烷-3α,17β-二醇(缩写5β-diol)、孕二醇(缩写PD)购自Sigma公司。2. 样品两名健康志愿者,一名男性40岁,尿样为sample 1;一名女性38岁,尿样为sample 2,均没有服用任何药物。收集其晨尿为阴性对照尿。阳性尿样为世界反兴奋剂机构水平考试所用尿样来自兴奋剂检测中心。3. 仪器[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]/燃烧炉/同位素比值质谱仪(HP6890/DELTA PLUS,Finnigan);高效液相色谱仪(Waters2796,检测器:Waters2996 PAD,自动收集器:Waters Fraction Collector Ⅲ);Anilent 5973i[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-质谱联用仪。4. 方法4.I [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/C/IRMS操作条件色谱柱:HP5毛细管色谱柱,25m×0.2mm i.d.×0.3μm;柱流速:1mL/min(室温);升温程序:60 ℃(2min)一50 ℃/min→255℃一2.5℃/min→280℃(6.5min);进样口温度:260℃;燃烧炉温度:960℃;质谱离子源:EI;参考气:CO2,1.8V。4.2 高效液相色谱仪操作条件色谱柱:ZQRBAX SB-C18(4.6mm×250mm,5μm);流动相:水-乙睛,梯度洗脱(0-18min:乙睛从30%→100%);流速1mL/min;柱温:室温。4.3 [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/MSD操作条件色谱柱:HP-1毛细管色谱柱,25 m×0.2mm i.d.×0.11μm;柱流速:1 mL/min (室温);升温程序:150 ℃(1min一5℃/min→200℃一30℃/min→310℃(10min)。4.4 样品预处理取尿样2mL,加人1Ml0.2mol pH=7.0的磷酸盐缓冲液和10μLβ-葡萄糖醛酸酶(5000 IU)混匀,在55℃恒温水浴中培养3h,取出后加pH=8.8的碳酸盐固体缓冲剂约100mg 和5mL叔丁基甲醚,振荡萃取,离心后,取出上层有机溶液,在加热的情况下,用氮气吹干,加人50μL甲醇溶解残渣,备用。将上述甲醇溶液置HPLC仪上,依前述色谱条件分离,分段收集流出液,确定收集时间程序。分别将流出组分用氮气吹干,加人50μL环己烷,备用。4.5 对照品溶液的制备分别配制对照品睾酮、雄酮、本胆烷醇酮、5α-雄烷-3α,17β-二醇、5β-雄烷-3α,17β-二醇,孕二醇的甲醇溶液,浓度为1mg/mL。4.6 样品测定4.6.1 [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/MSD 分析依上述[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/MSD仪器操作条件,取样品溶液及对照品溶液进行全扫描,扫描范围m/z 20~450,选择待测物的特征离子,获得SIM图。经对样品中与对照品有相同保留时间的峰进行质谱分析,及与标准品质谱图的对比,确定待测样品的组成。4.6.2 [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/C/IRMS分析依上述CC/C/IRMS仪器操作条件,对处理后的样品进行[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/C/IRMS分析,测得内源性类固醇激素的δ值。
质谱的检测限达到微克级别,是不是不太正常,最初怀疑是样本处理的不好造成检测的物质没有提取出来造成检测限偏大,但是后来拿来标准品进一下,发现ug/ml的标准品就几乎检测不到了,而且仪器的本底也随着检测的进行越来越高,另:检测的物质出峰圆钝,如何将峰高调好呀,现在出峰时间3-4分钟。仪器型号:Finnigan高效液相色谱-质谱联用仪,LC-10ADvp双泵,在线真空脱气机,恒温自动进样器,柱温箱,电喷雾离子化接口的四极杆质谱检测器
液相色谱-串联质谱法检测食品中维生素D含量 如何用液相色谱-串联质谱法(LC-MS/MS)检测食品中的维生素D含量。这项技术可是现代食品分析中的佼佼者,能够帮助我们精准地掌握食品中的营养信息。 样品前处理 提取:首先,我们需要从食品样品中提取维生素D。这一步很关键,因为提取效率直接影响最终的检测结果。通常,我们会使用有机溶剂,比如乙腈或甲醇,来提取维生素D。 净化:提取后的样品往往含有很多杂质,这些杂质会影响检测结果。因此,我们需要对提取液进行净化处理。常用的净化方法有固相萃取(SPE)和液液萃取(LLE)。 浓缩:净化后的样品溶液需要进行浓缩,以提高维生素D的浓度。常用的浓缩方法有氮气吹干和旋转蒸发。 仪器操作 流动相选择:选择合适的流动相对分离效果至关重要。通常,我们会使用水和有机溶剂(如甲醇或乙腈)的混合物作为流动相,并根据需要添加少量酸或缓冲液。 色谱柱选择:选择适合的色谱柱也很关键。C18反相色谱柱是常用的选择,因为它对维生素D有很好的保留效果。 质谱条件:设置合适的质谱条件,包括离子源温度、喷雾电压、碰撞能量等。这些参数的优化可以大大提高检测灵敏度和特异性。 故障排除 峰形不好:如果发现峰形不好,可能是由于流动相比例不合适或色谱柱污染。尝试调整流动相比例或清洗色谱柱。 灵敏度低:如果灵敏度不够,可能是由于样品提取效率低或仪器参数设置不当。检查提取方法并优化仪器参数。 杂峰干扰:如果出现杂峰干扰,可能是由于样品净化不彻底或流动相选择不当。尝试改进净化方法或更换流动相。 仪器故障:遇到仪器故障时,首先要保持冷静,然后根据仪器的报错信息查找原因。必要时,可以联系仪器厂家进行维修。 总之,液相色谱-串联质谱法检测食品中维生素D含量是一项复杂但非常重要的技术。通过掌握这些操作要点和故障排除方法,我们可以更加准确、高效地完成检测任务。
前段时间waters在本版发了一个关于“Waters将在10月7日推出什么新产品?”,相信很多版友都挺好奇的,现在谜底终于揭晓了,是ACQUITY QDa质谱检测器,我们来看看仪器介绍上说的:借助ACQUITY QDa质谱检测器,您可以:利用更高质量的质谱定性分析数据来有效补充沃特世光学检测器的定量分析数据,对成分进行准确鉴定。扩展现有PDA检测器的样品检测能力,对UV无响应的化合物以及光学检测不适合或是无法确定的化合物进行定量分析。您可以通过ACQUITY QDa质谱检测器获得信息量极其丰富的质谱数据。ACQUITY QDa质谱检测器如同光学检测器一样直观易用,并且能够稳定处理所有分析。同时,它能与您的色谱分析系统完美兼容,且仪器经过预先优化,适用于任何样品;而且无需像传统质谱仪那样要求用户针对不同样品的特异性进行仪器调谐。按上面的意思是ACQUITY QDa质谱检测器无需对仪器及样品进行任何参数的调谐优化,更或者说这种检测器根本就不需要这些参数,那ACQUITY QDa质谱检测器到底是质量分析器呢?还是只是光学检测器的一种升级版呢?还是其他的?它的检测限到底能达到多低的痕量呢?能达到目前主流的MS/MS的水平吗?您对ACQUITY QDa质谱检测器又是怎么看待的呢?原文由 victorlpyj(victorlpyj) 发表:关于这款检测器,我写了一篇报道,解读了相关参数和应用。详见:http://www.instrument.com.cn/news/20131028/115918.shtml。
请问各位怎么看AB质谱的检测器,和怎么调AB质谱检测器的数值?http://simg.instrument.com.cn/bbs/images/brow/emyc1004.gif
[color=#444444]质谱可以检测重烃吗,要检测煤层气中C2以上的烃[/color][color=#444444]我理解的是,只要是[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]能分开,质谱就能检测,对吗??[/color][color=#444444]TCD FID能做到的,质谱就能做的到,对吗??[/color]
质谱如何检测
气象色谱 质谱联用检测东湖水中的酚类物质 论文 谁可以给我建议呀
[color=#333333]质谱检测属于体外检测的什么类型?[/color]
质谱调谐发现水和氮气都在20左右,氧气5左右,随后加大分流比吹了半小时后,发现氮气达到了40,水和氧气也都高了很多,有没有遇到过这个问题的朋友,是怎么解决的
请问质谱检测时,检测条件设置不同,所测物质的质谱图有显著地不同吗?
想问一下大家为什么液相质谱需要用氮气?从哪里能得到详细的原理吗?谢谢!
检测氮气、甲烷和丙烷,用什么色谱柱,进样量是多少?氮气8%甲烷47%丙烷45%谢谢!都用TCD检测器
求气相色谱检测氢气、氧气、氮气色谱柱及最优实验条件?请高手赐教!
四级杆质谱仪是通过质量过滤后,怎么通过检测器来得到每种气体的含量的?是不是质谱仪在每次的分析时间中,会把样气中的每种气体都要分析出来,然后计算出它的比值,从而得到每种气体的含量的?能否介绍一下过滤后检测原理的详细过程?
各位老师好,请教一下,[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]质谱检测SIM检测时,标准曲线做的可以,但把标准曲线中的一个点当成样品,测定值减半,是什么原因呢?比如走一个100ng/ml的,检测出来只有40多。
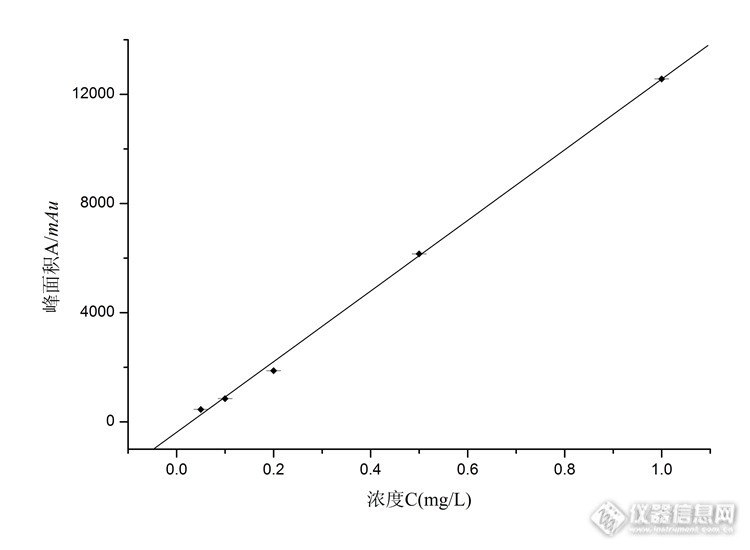
气相色谱-串联质谱技术检测水中己烯雌酚残留量摘要:本实验建立水中己烯雌酚残留量的气相色谱-串联质谱联用(GC-MS/MS)分析方法。样品采用乙酸乙酯提取,浓缩后甲醇溶解,经氮气吹干后加入BSTFA/TMCS衍生化试剂70℃衍生30mins,氮气吹干后甲醇定容待测,采用GC-MS/MS分析时,己烯雌酚衍生物在15 mins内流出。对采集的水样进行两种浓度的加标质控,回收率均在90%以上,相对标准偏差(RSD)在3.0%以内。在0.05~1.00 mg/L的浓度范围内呈线性关系,相关系数大于0.999。检出限为1.20 μg/L。该方法可准确用于环境水样中己烯雌酚残留的定量分析,灵敏度、精密度和准确度均满足残留检测的分析要求。关键词: 己烯雌酚;气相色谱-串联质谱;残留量;水中图分类号: 文献标识码: 文章编号: Determination of Diethylstilbestrol residues in water sample by gas chromatography-tandem mass spectrometry ABSTRACT: To establish a method determination of diethylstilbestrol (DES) in water sample by gas chromatography-tandem mass spectrometry (GC-MS/MS). The samples were extracted by ethyl acetate, derived with silanization reagent at 70℃ for 30 mins, then the extracts were determined with MS/MS detector based on SCAN and MRM mode, DES was well separated within 15 min. When the spiked average recoveries above 85%. The RSD ranged within 3.0%, The calibration of those pesticides were linear (correlation coefficients not less than 0.999) within the range of 0.05~1.00 μg/mL. The method was accurate and sensitive, was suitable for analysis of DES residues in water samples.KEY WORDS: diethylstilbestrol; gas chromatography-tandem mass spectrometry; residues; water sample1 引言 己烯雌酚(Diethylstilbestrol, DES)属激素类化合物,作为生长促进剂被广泛添加在饲料中应用于水产养殖行业促进鱼类生长,为水产业产品数量的增长发挥了一定作用。但己烯雌酚作为一种人工合成的具有雌激素样作用的化学物质,许多科学实验证实对人类和动物具有危害,导致机体代谢紊乱、发育异常或肿瘤等,是一种致癌物质,被欧盟等国在食用性动物饲养中列为禁用品,2002 年我国农业部发布176号公告禁止在畜禽水产养殖过程中使用己烯雌酚。己烯雌酚是脂溶性物质,难降解,易在动物体内残留,在水源和土壤中容易大量富积,造成环境激素污染恶性循环,由于会对环境水造成严重影响, 可通过饮用水威害人体健康 。因此,建立水样中己烯雌酚残留量的分析方法具有重要意义。目前,测定雌酚类激素的方法有酶联免疫(ELISA)法、比色法、电化学法、放射免疫(RIA) 法、色谱-质谱法、高效液相色谱(HPLC)法及毛细管电色谱法等。比色法、电化学法由于不能用于分离且干扰大等缺点,很少用于残留检测。本实验采用灵敏度和准确度较高的串联质谱检测器,可提高己烯雌酚定性与定量的准确性,二级质谱相对于一级质谱,其抗干扰能力更强,可有效消除单级质谱离子信息少的问题,对样品前处理的要求较低,可准确反映样品中己烯雌酚的残留量,其灵敏度、稳定性、准确度均较高。2 材料与方法2.1 材料2.1.1 试验原材料无公害水产养殖基地:淡水养殖用水。2.1.2 主要试剂与材料乙酸乙酯、二氯甲烷、正己烷、甲醇(色谱纯,美国Fisher Scientific公司);乙醚、叔丁基甲醚(优级纯,天津化学试剂厂);己烯雌酚标准品(德国Dr. Ehrenstorfor公司);衍生化试剂:N-甲基-N-三甲基硅基三氟乙酰胺(MSTFA)、二硫赤藓糖醇(DTE)、三甲基碘硅烷(TMIS)、N,O-双(三甲基硅烷基)三氟乙酰胺/三甲基氯硅烷(BSTFA/TMCS体积比99:1)(美国Supelco公司)。2.1.3 主要仪器气相色谱-串联质谱仪(7890B-7000B,美国Agilent公司);离心机(CT18RT,上海天美公司);氮吹仪(MG-2000,日本EYELA公司);旋转蒸发仪(SB-1100,日本EYELA公司)。2.2 标准溶液和衍生化试剂配制己烯雌酚标准溶液:配制10.0 mg/L的标准储备液,保存于1~4 ℃冰箱中冷藏,使用时用甲醇稀释至相应浓度做标准工作曲线。MSTFA-DTE-TMIS衍生化溶液:准确称取0.0040g DET溶解到2.00mL MSTFA,加入5.00μL TMIS,低温于1~4 ℃冰箱中冷藏避光保存。2.3 样品前处理2.3.1 分离提取准确量取40.0 mL水样品于具塞离心管中,加入20 mL提取试剂乙酸乙酯,振荡后放入高速离心机10000 r/min离心3 min,将上层有机相倒入浓缩瓶中,40℃减压浓缩近干,用甲醇溶液5 mL定容,转移至10 mL试管中氮气吹干。2.3.2 衍生化在吹干的试管中加入0.20 mL的衍生化试剂,在70℃烘箱中衍生化30 min,取出后氮气吹干1.00 mL甲醇定容上机。2.3.3 标准曲线及质控样分别配置0.05、0.10、0.20、0.50、1.00 mg/L浓度的标准工作液,按2.3.2衍生化后进样制作标准曲线,同时将样品添加不同浓度的标准品按2.3.1和2.3.1试验过程进行回收率质控试验。2.4 仪器条件色谱柱:HP-5MS(30 m×0.25 mm, 0.50 μm);载气:氦气(99.999%);恒流模式流速:1.00 mL/min;进样:1.00 μL,不分流;进样口温度:250 ℃;程序升温:80 ℃保持1 min,以20 ℃/min升温至280 ℃,保持10 min;离子化方式:电子轰击(EI);离子化能量:70 eV;离子源温度:230 ℃;传输线温度270 ℃;溶剂延迟:3 min;扫描方式:多反应监测(MRM)。扫描范围:50~450 amu;定量方式:外标法。3 结果与讨论3.1 提取溶剂选择本实验选择了乙醚、叔丁基甲基醚、乙酸乙酯、正己烷、二氯甲烷等溶剂作为提取剂,通过对添加浓度为1.00 mg/L的样品分别进行萃取回收率测定,由图1可见乙酸乙酯的萃取效率最高,萃取回收率稳定并达到90%以上, 所以本方法选择乙酸乙酯作为提取溶剂。http://ng1.17img.cn/bbsfiles/images/2016/07/201607081537_599845_1634341_3.jpg图1不同溶剂提取效率Fig. 1 Effect of Different Solvents on Extraction Efficiency 3.2 衍生化试剂及衍生化温度的选择本次试验选择MSTFA-DTE-TMIS和BSTFA/TMCS(99:1)两种衍生化试剂,均能与己烯雌酚的羟基进行硅烷化衍生反应,确定衍生化时间为30min,考察不同温度对于衍生化效率的影响,结果见图2。由图2可见,在70℃条件下衍生化效率最好,不同温度条件下衍生化试剂BSTFA/TMCS (99:1)的效率均优于MSTFA-DTE-TMIS,同时BSTFA/TMCS(99:1)有配制好的商品可供购买,使用便捷,且毒性也较小,因此本次试验选用BSTFA/TMCS为最佳衍生化试剂。[img=,
想做核酸检测,但因为是新手,所以N多问题都不是很懂,还望各位大神不吝赐教!1、用质谱对核酸进行定性分析,这样的方法可行吗?2、如果可行,那是不是就是将我做出的质谱图拿来与质谱数据库中的该核酸标准质谱图进行对比,以确定我所得出的就是我想检测的核酸呢?这个不知道思路是不是这样的,如果不是,还望帮我指条明路http://assets.dxycdn.com/gitrepo/bbs/emotions/default/078.gif 如果是这样思路的话,那标准质谱图/质谱数据库我该去哪儿找呢?
[color=#444444]利用液相色谱-质谱做的酯类润滑油,检测出分子量都在七八百,实际情况应该在四百左右。请问什么因素会导致检测结果大这么多呢?[/color]
[color=#444444]求助关于质谱检测条件的问题。中药材提取物,在[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]测定时,极性较大的物质,为什么在质谱中响应很小,甚至离子流图基本看不到信号,而极性小的物质可以看到信号。如何改善条件呢?(流动相为乙腈-0.1%甲酸水梯度洗脱。)[/color]







 我要推广仪器
我要推广仪器
 下载APP
下载APP