在生物大分子纯化分析特别是蛋白质纯化分析中,色谱是非常重要而且常用的一种技术。 一、凝胶过滤 凝胶过滤又叫分子筛色谱,其原因是凝胶具有网状结构,小分子物质能进入其内部,而大分子物质却被排除在外部。当一混合溶液通过凝胶过滤色谱柱时,溶液中的物质就按不同分子量筛分开了。 二、离子交换色谱 离子交换色谱是在以离子交换剂为固定相,液体为流动相的系统中进行的。离子交换剂是由基质、电荷基团和反离子构成的。离子交换剂与水溶液中离子或离子化合物的反应主要以离子交换方式进行,或借助离子交换剂上电荷基团对溶液中离子或离子化合物的吸附作用进行。 三、吸附色谱 1、 吸附柱色谱 吸附柱色谱是以固体吸附剂为固定相,以有机溶剂或缓冲液为流动相构成柱的一种色谱方法。 2、 薄层色谱 薄层色谱是以涂布于玻板或涤纶片等载体上的基质为固定相,以液体为流动相的一种色谱方法。这种色谱方法是把吸附剂等物质涂布于载体上形成薄层,然后按纸色谱操作进行展层。 3、 聚酰胺薄膜色谱 聚酰胺对极性物质的吸附作用是由于它能和被分离物之间形成氢键。这种氢键的强弱就决定了被分离物与聚酰胺薄膜之间吸附能力的大小。色谱时,展层剂与被分离物在聚酰胺膜表面竞争形成氢键。因此选择适当的展层剂使分离在聚酰胺膜表面发生吸附、解吸附、再吸附、再解吸附的连续过程,就能导致分离物质达到分离目的。 四、 亲和色谱 亲和色谱是根据生物大分子和配体之间的特异性亲和力,将某种配体连接在载体上作为固定相,而对能与配体特异性结合的生物大分子进行分离的一种色谱技术。亲和色谱是分离生物大分子最为有效的色谱技术,分辨率很高。 亲和色谱的原理与众所周知的抗原一抗体、激素一受体和酶一底物等特异性反应的机理相类似,每对反应物之间都有一定的亲和力。正如在酶与底物的反应中,特异的废物(S'')才能和一定的酶(E)结合,产生复合物(E-S'')一样。在亲和色谱中是特异的配体才能和一定的生命大分子之间具有亲和力,并产生复合物。而亲和色谱与酶一底物反应不同的是,前者进行反应时,配体(类似底物)是固相存在;后者进行反应时,底物呈液相存在。实质上亲和色谱是把具有识别能力的配体L(对酶的配体可以是类似底物、抑制剂或辅基等)以共价键的方式固化到含有活化基团的基质M(如活化琼脂糖等)上,制成亲和吸附剂M-L,或者叫做固相载体。而固化后的配体仍保持束缚特异物质的能力。 因此,当把围相载体装人小色谱柱(几毫升到几十毫升床体积)后,让欲分离的样品液通过该柱。这时样品中对配体有亲和力的物质S就可借助静电引力、范德瓦尔力,以及结构互补效应等作用吸附到固相载体上,而无亲和力或非特异吸附的物质则被起始缓冲液洗涤出来,并形成了第一个色谱峰。然后,恰当地改变起始缓冲 液的PH值、或增加离子强度、或加人抑③剂等因子,即可把物质S从固相载体上解离下来,并形成了第M个色谱峰(见图6-2)。显然,通过这一操作程序就可把有效成分与杂质满意地分离开。如果样品液中存在两个以上的物质与固相载体具有亲和力(其大小有差异)时,采用选择性缓冲液进行洗脱,也可以将它们分离开。用过的固相载体经再生处理后,可以重复使用。 上面介绍的亲和色谱法也是特异性配体亲和色谱法。另外还有通用性配体亲和色谱法。这两种亲和色谱法相比,前者的配体一般为复杂的生命大分子物质(如抗体、受体和酶的类似底物等),它具有较强的吸附选择性和较大的结合力。而后者的配体则一般为简单的小分子物质(如金属、染料,以及氨基酸等),它成本低廉、具有较高的吸附容量,通过改善吸附和脱附条件可提高色谱的分辨率。 五、聚焦色谱 聚焦色谱也是一种柱色谱。因此,它和另外的色谱一样,照例具有流动相,其流动相为 多缓冲剂,固定相为多缓冲交换剂。 聚焦色谱原理可以尝试从PH梯度溶液的形成、蛋白质的行为和聚焦效应三方面来阐述。 1、PH梯度溶液的形成 在离子交换色谱中,PH梯度溶液的形成是靠梯度混合仪实现的。例如,当使用阴离子 剂进行色谱时,制备PH由高到低呈线性变化的梯度溶液的方法是,在梯度仪的混合室(这色谱柱者)中装高PH溶液,而在另一室装低PH极限溶液,然后打开色谱柱的下端出口,让洗脱液连续不断地流过柱体。这时从柱的上部到下部溶液的PH值是由高到低变化的。而在聚焦色谱中,当洗脱液流进多缓冲交换剂时,由于交换剂带具有缓冲能力的电荷基团,故PH梯度溶液可以自动形成。 2.蛋白质的行为 蛋白质所带电荷取决于它的等电点(PI)和色谱柱中的PH值。当柱中的PH低于蛋白质的PI时,蛋白质带正电荷,且不与阴离于交换剂结合。而随着洗脱剂向前移动,固定相中的PH值是随着淋洗时间延长而变化的。当蛋白质移动至环境PH高于其PI时,蛋白质由带正电行变为带负电荷,并与阴离子交换剂结合。由于洗脱剂的通过,蛋白质周围的环境PH 再次低于PI时,它又带正电荷,并从交换剂解吸下来。随着洗脱液向柱底的迁移,上述过程将反复进行,于是各种蛋白质就在各自的等电点被洗下来,从而达到了分离的目的。 不同蛋白质具有不同的等电点,它们在被离子交换剂结合以前,移动之距离是不同的,洗脱出来的先后次序是按等电点排列的。
要正确地选择色谱分离方法,首先必须尽可能多的 了解样品的有关性质,其次必须熟悉各种色谱方法的主要特点及其应用范围。选择色谱分离方法的主要根据 是样品的相对分子质量的大小,在水中和有机溶剂中的溶解度,极性和稳定程度以及化学结构等物理、化学性质。一、相对分子质量对于相对分子质量较低(一般在200以下),挥发性比较好,加热又不易分解的样品,可以选择[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法进行分析。相对分子质量在200 ~ 2000的化合物,可用液固吸附、液-液分配和离子交换色谱法。相对分子质量高于2000,则可用空间排阻色谱法。二、溶解度水溶性样品最好用离子交换色谱法和液液分配色谱法;微溶于水,但在酸或碱存在下能很好电离的化合物,也可用离子交换色谱法;油溶性样品或相对非极性的混合物,可用液-固色谱法。三、化学结构若样品中包含离子型或可离子化的化合物,或者能与离子型化合物相互作用的化合物(例如配位体及有机螯合剂),可首先考虑用离子交换色谱,但空间排阻和液液分配色谱也都能顺利地应用于离子化合物;异构体的分离可用液固色谱法;具有不同官能团的化合物、同系物可用液液分配色谱法;对于高分子聚合物,可用空间排阻色谱法。
有哪些常用的色谱分离方法???
你模式过色谱条件吗?你测定过多个组分吗?来分享下你使用离子色谱时改善分离度方法的方法吧,看看你都是怎么改善的!!!!
[color=#444444][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]6820型号,柱子是TD-5,主要分离俩个反应体系,一个是苯甲醛和丙二睛在甲苯中的 反应,另一个是苯甲醛和氰乙酸乙酯在甲苯中的反应,或者这俩种反应在水或者乙醇中的反应, 怎样建立方法 能将其分离开?谢谢了[/color]
当色谱峰出现重叠,改变硬件条件的情况下如果还不能解决。 可以用卡尔曼滤波算法进行软件分离而得到准确的面积。http://ng1.17img.cn/bbsfiles/images/2017/03/201703071038_01_1780790_3.png上图是一个分离效果图。通过众多的样品测试分析,卡尔曼滤波算法对于重叠程度低于90%的,分离效果相当好,对于重叠程度大于90%而小于95%的有一定误差,而超过95%的基本就无能为力了。色谱分析中经常出现基线漂移,出峰时间是变动的。这也可以通过采用一定软件算法进行校正。在校正前需要指定一个正常的谱,然后通过神经网络算法进行学习。测试其它样品时如果发现有漂移就会计算出漂移距离(时间),再进行校正。以上重叠峰分离方法和漂移校正方法同样适合光谱分析。如果需要技术交流的,可点击头像联系
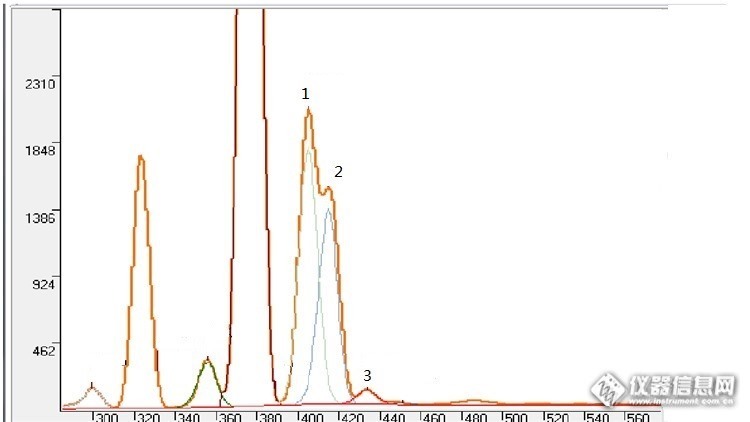
当色谱峰出现重叠,改变硬件条件的情况下如果还不能解决。可以用卡尔曼滤波算法进行软件分离而得到准确的面积。http://ng1.17img.cn/bbsfiles/images/2017/03/201703071038_01_1780790_3.png上图是一个分离效果图。通过众多的样品测试分析,卡尔曼滤波算法对于重叠程度低于90%的,分离效果相当好,对于重叠程度大于90%而小于95%的有一定误差,而超过95%的基本就无能为力了。色谱分析中经常出现基线漂移,出峰时间是变动的。这也可以通过采用一定软件算法进行校正。在校正前需要指定一个正常的谱,然后通过神经网络算法进行学习。测试其它样品时如果发现有漂移就会计算出漂移距离(时间),再进行校正。以上重叠峰分离方法和漂移校正方法同样适合光谱分析。如果需要技术交流的,可联系我 电话:13926563756 qq:648048428 Email: midstone@126.com
高效色谱仪分离方法的选择原则:一、根据相对分子质量选择:1、相对分子质量很低的样品采用气相色谱。2、液液分配色谱、液固吸附色谱和离子交换色谱最适合分析相对分子质量为200~2000的样品。3、相对分子质量大于2000的样品,采用凝胶色谱为最优。二、根据溶解度选择:1、溶于水并能离解的样品,采用离子交换色谱。2、溶于烃类(如苯或异辛烷等)的样品,可采用液固吸附色谱。3、溶于CCl4的样品,多采用液液分配色谱和液固吸附色谱。4、既溶于水又溶于异丙醇的样品,常用水和异丙醇的混合液作液液分配色谱的流动相,以疏水性化合物作固定相。三、根据分子结构选择:1、酸、碱化合物采用离子交换色谱。2、脂肪族和芳香族采用液液分配色谱、液固吸附色谱。3、异构体采用液固吸附色谱。4、同系物不同官能团和强氢键化合物采用液液分配色谱。
近年来离子色谱研究的一个重要趋势是研究各种分离效率高, 选择性好, 分析速度快, 可同时分析阴离子和阳离子的色谱柱. 研究的重点是将涂覆有生物表面活性剂的物质作离子色谱固定相, 并已在光学异构体和无机离子分离分析方面展示出独特的优越性和发展潜力. 1994年, Hu Wezhi等人首先采用在一分子内含有正负电荷的两性离子分子的表面活性剂作色谱固定相, 开创了静电离子色谱法. 本文利用自制的静电离子色谱柱, 选用不同种类流动相, 对含有不同阴离子的钠盐进行分离, 并初步探讨在磁场中静电离子色谱的保留行为. 1 实验部分 1.1 仪器和试剂 LC-4A高效液相色谱仪; RID-2AS示差析光检测器, C-R2A数据处理机. 静电离子色谱柱(自制), 流动相分别为水, 10 mmol/L Na2HPO4-NaH2PO4缓冲液(pH=6.8), 2.4 mmol/L NaHCO3和3 mmol/L Na2CO3; 1 mmol/L十二烷基磺酸钠. 所用试剂均为优级纯或分析纯; 溶液用二次蒸馏水按常规配制. 1.2 色谱柱制备和分离方法 把含有胆汁酸盐水溶液通过动态涂层法涂覆在ODS表面. 选用国产ODS分离柱(4.6 mm×250 mm), 将30 mmol/L的CHAPS溶液(经0.4 μm滤膜过滤)以 0.7 mL/min流速流经ODS柱80 min, 收集流出液重复上述操作2次, 然后用水冲洗40 min, 即得到在ODS柱表面涂覆一层含有正/负电荷胶束的静电离子色谱柱. 静电离子色谱法是利用在ODS载体上涂覆在同一分子内同时含有正/负两种电荷的胆汁酸诱导体胶束作固定相, 纯水或电解质溶液作流动相, 被测样品中的阴离子和阳离子通过纯粹的静电吸引、 离子配对后形成正、 负离子的缔合物(离子对), 由于被测离子的电荷和半径、 离子种类和离子浓度的不同, 因此形成的各种离子对受涂覆在固定相上的表面活性剂所带的正/负电荷静电吸引和排斥作用力不同而相互分离. 分离后的离子对进入检测器进行定量检测. 实验表明, 用本法制备的静电离子色谱柱, 连续使用3个月未发现分离效率下降. 2 结果与讨论 2.1 流动相和色谱图 分别以纯水、磷酸盐缓冲溶液为流动相得到色谱分离图 纯水为流动相时, Na2SO4和NaBr, KNO3和NaNO3, Na2S2O3和NaF+NaNO3各离子对得到分离, 但NaF与NaNO3不能分离开. 而磷酸盐为缓冲溶液时(图2), 不但Na2SO4和NaBr得到分离, 而且Na2S2O3, NaF, NaNO3也可相互分离. 由图2可见, 与纯水流动相相比, 流动相中磷酸盐的存在使各离子对保留时间和色谱峰形状发生变化, 虽然各离子对保留时间显著增加, 但出峰顺序未发生变化. 实验表明, 各离子对的保留时间与阴阳离子的半径、 电荷、 流动相种类和离子强度有关, 在流动相中加入不同种类的电解质溶液将有利于某些离子对的分离. 分别以碳酸盐、十二烷基磺酸钠为流动相得到的静电离子色谱分离图如图3所示. 由图3可见NaBr和Na2SO4可以完全分离, 与纯水为流动相相比, NaBr和Na2SO4的分离效率提高, 但保留时间增加. 特别是以十二烷基磺酸钠(表面活性剂)为流动相时, 使NaBr的保留时间延长(见图3(b)), 这说明表面活性剂的存在将对离子对的分离效率产生重要影响. 可以认为, 在流动相中加入电解质溶液, 除样品离子与固定相相互作用外, 流动相中电解质也参与了与固定相之间的静电吸引和排斥作用, 由于各离子对和电解质与固定相相互竞争的静电作用, 提高了各离子对的分离度. 2.2 流动相流速影响 当流动相流速不同时, 各离子对的保留时间发生改变. 纯水为流动相时, NaBr和Na2SO4离子对的保留时间与纯水流速的关系. 实验表明, 当采用不同种类流动相时, 随着流动相流速的增加, 保留时间都有不同程度的缩短. 但要根据被分离的离子对的分离效率和分析速度来选择流动相流速, 本实验选择流动相流速为0.6 mL/min. 2.3 外加磁场对静电离子色谱分离的影响 将静电离子色谱置于静态磁场(Nb磁铁, 160 mm×30 mm)中, 考察各离子对的分离效率和保留时间. 实验表明, 在外加磁场作用下, 纯水为流动相时, NaNO3和Na2S2O3离子对的保留时间稍向后位移(见图5), 但二者的峰形状未发生变化. 这可能是在离子对形成和洗脱过程中, 由于外加磁场的作用, 使形成的离子对与涂覆在载体上胆汁酸盐胶束所带的正负电荷静电吸引和排斥作用力发生变化, 打破了原来的平衡状态, 使离子对的保留时间发生位移.
色谱分离要求在最短的时间内,以“塞子”形式打进一定量的试样,进样方法可分为:1.气体试样:大致进样方法有四种:(1)注射器进样(2)量管进样(3)定体积进样(4)气体自动进样。一般常用注射器进样及气体自动进样。注射器进样的优点是使用灵活,方法简便,但进样量重复性较差。气体自动进样是用定量阀进样,重复性好,且可自动操作。
两种标准品,色谱峰有重叠,请问有什么方法可以分离吗?我不想改变流动相的配比,改变流速是不是可以呢?[em0906]柱温35,254nm,流动相甲醇:乙酸=45:0.2%
色谱分离要求在最短的时间内,以“塞子”形式打进一定量的试样,进样方法可分为:液体试样:一般用微量注射器进样,方法简便,进样迅速。也可采用定量自动进样,此法进行重复性良好。固体试样:通常用溶剂将试样溶解,然后采用和液体进样同样方法进样。也有用固体进样器进样的。
求助液相色谱分离甲酸甲醇乙酸乙醇方法,分不开甲酸和乙醇,现在流动相是磷酸二氢钠和乙腈,C18色谱柱
1、稀释样品 对组成复杂的样品,若待测离子对树脂亲合力相差颇大,就要作几次进样,并用不同浓度或强度的淋洗液或梯度淋洗。对固定相亲合力差异较大的离子,增加分离度的最简单方法是稀释样品或作样品前处理。例如盐水中SO2-4和Cl-的分离。若直接进样,其色谱峰很宽而且拖尾,表明进样量已超过分离柱容量,在常用的分析阴离子的色谱条件下,30min之后Cl-的洗脱仍在继续。在这种情况下,在未恢复稳定基线之前不能再进样。若将样品稀释10倍之后再进样就可得到Cl-与痕量SO2-4之间的较好分离。对阴离子分析推荐的最大进样量,一般为柱容量的30%,超过这个范围就会出现大的平头峰或肩峰 2、改变分离和检测方式 若待测离子对固定相亲合力相近或相同,样品稀释的效果常不令人满意。对这种情况,除了选择适当的流动相之外,还应考虑选择适当的分离方式和检测方式。例如,NO-3和ClO-3,由于它们的电荷数和离子半径相似,在阴离子交换分离柱上共淋洗。但ClO-3的疏水性大于NO-3,在离子对色谱柱上就很容易分开。又如NO-2与Cl-在阴离子交换分离柱上的保留时间相近,常见样品中Cl-的浓度又远大于NO-2,使分离更加困难,但NO-2有强的UV吸收,而Cl-则很弱,因此,应改用紫外作检测器测定NO-2,用电导检测Cl-,或将两种检测器串联,于一次进样同时检测Cl-与NO-2。对高浓度强酸中有机酸的分析,若采用离子排斥,由于强酸不被保留,在死体积排除,将不干扰有机酸的分离。 3、样品前处理 对高浓度基体中痕量离子的测定,例如海水中阴离子的测定,最好的方法是对样品作适当的前处理。除去过量Cl-的前处理方法有:使样品通过Ag+型前处理柱除去Cl-,或进样前加AgNO3到样品中沉淀Cl-;也可用阀切换技术,其方法是使样品中弱保留的组分和90%以上的Cl-进入废液,只让10%左右的Cl-和保留时间大于Cl-的组分进入分离柱进行分离。对含有大的有机分子的样品,应于进样前除去有机物,较简单的方法是用Dionex的前处理柱OnGuard的RP或P柱或在线阀切换除去有机基体。 4、选择适当的淋洗液 离子色谱分离是基于淋洗离子和样品离子之间对树脂有效交换容量的竞争,为了得到有效的竞争,样品离子和淋洗离子应有相近的亲合力。下面举例说明选择淋洗液的一般原则。用CO2-32HCO-作淋洗液时,在Cl-之前洗脱的离子是弱保留离子,包括一价无机阴离子、短碳链一元羧酸和一些弱离解的组分,如F-,甲酸,AsO-2,CN-和S2-等。对乙酸,甲酸与F-、Cl-等的分离应选用较弱的淋洗离子,常用的弱淋洗离子有HCO-3,OH-和B4O2-7。由于HCO-3和OH-易吸收空气中CO2,CO2在碱性溶液中会转变成CO2-3,CO2-3之淋洗强度较HCO-3和OH-大,因而不利于上述弱保留离子的分离。B4O2-7亦为弱淋洗离子,但溶液稳定,是分离弱保留离子的推荐淋洗液。中等强度的碳酸盐淋洗液对高亲和力组分的洗脱效率低。 对离子交换树脂亲合力强的离子有两种情况,一种是离子的电荷数大,如PO3-4,AsO3-4和多聚磷酸盐等;一种是离子半径较大,疏水性强,如I-,SCN-,S2O2-3,苯甲酸和柠檬酸等。对前者以增加淋洗液的浓度或选择强的淋洗离子为主。对后一种情况,推荐的方法是在淋洗液中加入有机改进剂(如甲醇、乙腈和对氰酚等)或选用亲水性的柱子,有机改进剂的作用主要是减少样品离子与离子交换树脂之间的非离子交换作用,占据树脂的疏水性位置,减少疏水性离子在树脂上的吸附,从而缩短保留时间,减少峰的拖尾,并增加测定灵敏度。 在离子色谱中,可由加入不同的淋洗液添加剂来改善选择性,这种淋洗液添加剂只影响树脂和所测离子之间的相互作用,而不影响离子交换。对与树脂亲合力较强的离子,如一些可极化的离子,I-和ClO4-,以及疏水性的离子,苯甲酸和三乙胺等,在淋洗液中加入适量极性的有机溶剂如甲醇或乙腈,可缩短这些组分的保留时间并改善峰形的不对称性。为了减少样品离子与树脂之间的非离子交换作用,减少树脂对疏水性离子的吸附,在阴离子分析中,可在淋洗液中加入对氰酚。如测定1%NaCl中的痕量I-和SCN-时,加入对氰酚占据树脂对I-和SCN-的吸附位置,从而减少峰的拖尾并增加测定灵敏度。IC中,一价淋洗离子洗脱一价待测离子,二价淋洗离子洗脱二价待测离子,淋洗液浓度的改变对二价和多价待测离子保留时间的影响大于一价待测离子。若多价离子的保留时间太长,增加淋洗液的浓度是较好的方法。
1 稀释样品对组成复杂的样品, 若待测离子对树脂亲合力相差颇大, 就要作几次进样, 并用不同浓度或强度的淋洗液或梯度淋洗. 对固定相亲合力差异较大的离子, 增加分离度的最简单方法是稀释样品或作样品前处理. 例如盐水中SO2 -4 和Cl - 的分离. 若直接进样, 其色谱峰很宽而且拖尾, 表明进样量已超过分离柱容量, 在常用的分析阴离子的色谱条件下, 30min 之后Cl - 的洗脱仍在继续. 在这种情况下,在未恢复稳定基线之前不能再进样. 若将样品稀释10 倍之后再进样就可得到Cl - 与痕量SO2 -4 之间的较好分离. 对阴离子分析推荐的最大进样量, 一般为柱容量的30 % , 超过这个范围就会出现大的平头峰或肩峰2 改变分离和检测方式若待测离子对固定相亲合力相近或相同, 样品稀释的效果常不令人满意. 对这种情况, 除了选择适当的流动相之外, 还应考虑选择适当的分离方式和检测方式. 例如, NO-3 和ClO-3 , 由于它们的电荷数和离子半径相似, 在阴离子交换分离柱上共淋洗. 但ClO-3 的疏水性大于NO-3 , 在离子对色谱柱上就很容易分开. 又如NO-2 与Cl - 在阴离子交换分离柱上的保留时间相近, 常见样品中Cl - 的浓度又远大于NO-2 , 使分离更加困难, 但NO-2 有强的UV 吸收, 而Cl - 则很弱, 因此, 应改用紫外作检测器测定NO-2 , 用电导检测Cl - , 或将两种检测器串联, 于一次进样同时检测Cl - 与NO-2 . 对高浓度强酸中有机酸的分析, 若采用离子排斥, 由于强酸不被保留, 在死体积排除, 将不干扰有机酸的分离.3 样品前处理对高浓度基体中痕量离子的测定, 例如海水中阴离子的测定, 最好的方法是对样品作适当的前处理. 除去过量Cl - 的前处理方法有: 使样品通过Ag+ 型前处理柱除去Cl - , 或进样前加AgNO3 到样品中沉淀Cl - 也可用阀切换技术, 其方法是使样品中弱保留的组分和90 %以上的Cl - 进入废液, 只让10 %左右的Cl - 和保留时间大于Cl - 的组分进入分离柱进行分离. 对含有大的有机分子的样品, 应于进样前除去有机物, 较简单的方法是用Dionex 的前处理柱OnGuard 的RP 或P 柱或在线阀切换除去有机基体.4 选择适当的淋洗液[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]分离是基于淋洗离子和样品离子之间对树脂有效交换容量的竞争, 为了得到有效的竞争, 样品离子和淋洗离子应有相近的亲合力. 下面举例说明选择淋洗液的一般原则. 用CO2 -3 2HCO-作淋洗液时, 在Cl - 之前洗脱的离子是弱保留离子, 包括一价无机阴离子、短碳链一元羧酸和一些弱离解的组分, 如F- , 甲酸, AsO-2 , CN- 和S2 - 等. 对乙酸, 甲酸与F- 、Cl - 等的分离应选用较弱的淋洗离子, 常用的弱淋洗离子有HCO-3 , OH- 和B4O2 -7 . 由于HCO-3 和OH- 易吸收空气中CO2 , CO2 在碱性溶液中会转变成CO2 -3 , CO2 -3 之淋洗强度较HCO-3 和OH- 大, 因而不利于上述弱保留离子的分离. B4O2 -7 亦为弱淋洗离子, 但溶液稳定, 是分离弱保留离子的推荐淋洗液. 中等强度的碳酸盐淋洗液对高亲和力组分的洗脱效率低.对离子交换树脂亲合力强的离子有两种情况, 一种是离子的电荷数大, 如PO3 -4 , AsO3 -4 和多聚磷酸盐等 一种是离子半径较大, 疏水性强, 如I - , SCN- , S2O2 -3 , 苯甲酸和柠檬酸等. 对前者以增加淋洗液的浓度或选择强的淋洗离子为主. 对后一种情况, 推荐的方法是在淋洗液中加入有机改进剂(如甲醇、乙腈和对氰酚等) 或选用亲水性的柱子, 有机改进剂的作用主要是减少样品离子与离子交换树脂之间的非离子交换作用, 占据树脂的疏水性位置, 减少疏水性离子在树脂上的吸附, 从而缩短保留时间, 减少峰的拖尾, 并增加测定灵敏度.在[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]中, 可由加入不同的淋洗液添加剂来改善选择性, 这种淋洗液添加剂只影响树脂和所测离子之间的相互作用, 而不影响离子交换. 对与树脂亲合力较强的离子, 如一些可极化的离子, I-和ClO4- , 以及疏水性的离子, 苯甲酸和三乙胺等, 在淋洗液中加入适量极性的有机溶剂如甲醇或乙腈, 可缩短这些组分的保留时间并改善峰形的不对称性. 为了减少样品离子与树脂之间的非离子交换作用, 减少树脂对疏水性离子的吸附, 在阴离子分析中, 可在淋洗液中加入对氰酚. 如测定1 %NaCl中的痕量I- 和SCN- 时, 加入对氰酚占据树脂对I- 和SCN- 的吸附位置, 从而减少峰的拖尾并增加测定灵敏度. [url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]IC[/color][/url] 中, 一价淋洗离子洗脱一价待测离子, 二价淋洗离子洗脱二价待测离子, 淋洗液浓度的改变对二价和多价待测离子保留时间的影响大于一价待测离子. 若多价离子的保留时间太长, 增加淋洗液的浓度是较好的方法.
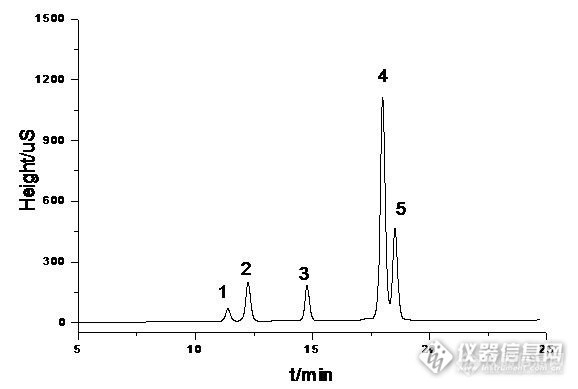
五种芳环磺酸盐的液相色谱分离方法浅谈 1-萘磺酸钠,2-萘磺酸钠,蒽醌1,5-二磺酸钠、蒽醌1,8-二磺酸钾和氨基萘酚-二磺酸钠五种物质均为强极性芳环磺酸盐化合物,在水溶液中完全电离成离子状态。在液相色谱分析中,五种物质的保留性质相似;因其为离子状态,C18反相柱对其没有保留,无法将其成功分离。本分析方法中采用离子对-反相色谱方法对其进行分离。具体分析条件和色谱分离图如下: 表1 色谱分析条件分析仪器U3000 HPLC 系统 色谱柱Diamonsil®(钻石)C18200*4.6mm,5um流动相A乙腈梯度洗脱:30minA: 20% ___ 45%B0.1%TBA+0.3%磷酸二氢钾pH6.5 温度30℃ 流速1mL/min 检测波长222nm http://ng1.17img.cn/bbsfiles/images/2016/09/201609141634_609777_3137073_3.jpg 图1 五种芳环磺酸盐的液相色谱分离图 1:蒽醌1,5-二磺酸钠,2:氨基萘酚-二磺酸钠,3:蒽醌1,8-二磺酸钾,4:1-萘磺酸钠,5:2-萘磺酸钠总结: 从色谱条件分析,本方法使用的是常规的反相C18色谱柱,在流动相中加入离子对试剂的方法增强保留性,用磷酸二氢钾调节pH至近中性。方法建立过程中发现pH和乙腈的比例对五种物质的分离影响较大,溶液的pH的大小影响芳环磺酸盐在水溶液中的分子形态,酸性溶液中,各物质形成成分子,中性和碱性条件下电离成离子状态,碱性溶液中有利于与离子对试剂的结合,但考虑到仪器和色谱柱不耐碱性能,选择中性范围较为合适。流动相中乙腈的比例对各物质在色谱柱中的保留时间影响。经过多次试验发现,梯度条件能较理想的将物种五种物质分开,而在等度条件下无法实现,特别是蒽醌1,5-二磺酸钠和氨基萘酚-二磺酸钠,1-萘磺酸那和2-萘磺酸钠两组性能保留性能相似的物质较难分开。 谱分离图来看,三种芳环二磺酸盐和萘磺酸盐完全分开,分离效果较好。蒽醌1,5-二磺酸钠和蒽醌1,8-二磺酸钾因空间结构差异,所表现出来的保留性有明显的区别,因此可以较好的分离。1-萘磺酸钠和2-萘磺酸钠结构性质非常相似,化学性质也相近,较难分离,目前还未建立将两者完全分离的方法。图中所示分离度不如其他物质理想。
各位有色谱方法开发经验的前辈:现有两个问题需请教1.对于高浓度主峰,峰宽很宽,有6分钟样子,在紧随其后有1杂质峰,与主峰分离度只有0.8,色谱条件除了色谱柱可以更换外,其他条件均不能改变,那么用何种色谱柱可以提高两者的分离度为1.5以上;2.哪类色谱柱更适合于分析高浓度化合物;谢谢各位赐教,不胜感激!
离子色谱仪分离是基于样品离子和淋洗离子之间对树脂有效交换容量的竞争,为了得到有效的竞争,样品离子和淋洗离子应有相近的亲和力。一、分离弱保留离子的淋洗液: 用CO32-或HCO3-作淋洗液时,在Cl-之前洗脱的离子是弱保留离子,包括一价无机阴离子、短碳链一元羧酸和一些弱离解的组分等。 对乙酸、甲酸、F-和Cl-等分离应选用较弱的淋洗离子,常用的弱淋洗离子有HCO3-、OH-和B4O72-。由于HCO3-和OH-易吸收空气中CO2,CO2在碱性溶液中会转变成CO32-,CO32-的淋洗强度比HCO3-和OH-大,不利于上述弱保留离子的分离。B4O72-为弱淋洗离子,且溶液稳定,是分离弱保留离子的推荐淋洗液。二、分离高亲和力离子的淋洗液: 中等强度的碳酸盐淋洗液对高亲和力离子的洗脱效率低。对离子交换树脂亲和力强的离子有两种情况:一种是离子电荷数大,如PO43-、AsO43-和多聚磷酸盐等。另一种是离子半径较大、疏水性强,如I-、SCN-、S2O32-、苯甲酸和柠檬酸等。三、分离电荷数大的离子的淋洗液: 以增加淋洗液的浓度或选择强的淋洗离子为主。四、分离半径较大和疏水性强的离子的淋洗液: 对半径较大和疏水性强的离子,推荐方法是在淋洗液中加入有机改进剂(如甲醇、乙腈和对氰酚等)或选用亲水性色谱柱。有机改进剂的作用主要是减少样品离子与离子交换树脂之间的非离子交换作用,占据树脂的疏水性位置,减少疏水性离子在树脂上的吸附,从而缩短保留时间,减少峰拖尾,并增加检测灵敏度。 离子色谱中,一价淋洗离子洗脱一价待测离子,二价淋洗离子洗脱二价待测离子,淋洗液浓度的改变对二价和多价待测离子保留时间的影响大于一价待测离子。若多价离子的保留时间太长,增加淋洗液的浓度是较好的方法。

[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分离氧气、氮气、一氧化碳、二氧化碳、丙烯、丙烷,什么色谱柱能分离,或者用什么方法把他们分离定量。求大神帮忙,小女子不胜感激....
1. 是不是所以的混合物均可用柱色谱分离?柱色谱是万能分离方法吗?2.含有金属物质的分离一般用什么样的溶剂较好?
分子大小组成相似,分子大小相近,如何用液相色谱法分离分级?液相色谱中哪种方法更好呢?
残留溶剂检查时,能用什么色谱柱或方法将乙醇和乙腈有效分离?谢谢!
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=171584]蛋白质的多维色谱分离[/url]蛋白质的多维色谱分离通常生物体内提取的蛋白质样品成分复杂,应用多维色谱进行分离的方法必将流行。文章简单介绍了多维色谱分离的理论,算是一个入门材料吧。
目的:建立一个[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]条件同时分离测定环孢素A中乙醇及丙二醇的含量。方法:以GDX-101为固定相,柱长为2 m,进样口温度为210 ℃,检测器为280 ℃,柱温采用程序升温,氮气为载气,以二甲基亚砜为溶剂,以正丙醇为内标。结果:乙醇及丙二醇进样量分别在2.0~6.0 μg,1.0~3.0 μg,其峰面积与浓度呈良好的线性关系,加样回收率分别为99.9%(RSD<0.8%,n=5),101.4%(RSD<1.1%,n=5),精密度良好。结论:此[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]条件可同时测定环孢素A中乙醇及丙二醇的含量,方法简便准确。关键词 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法 乙醇 丙二醇 环孢素A山地明(环孢素A)为诺华制药有限公司的产品,是一种免疫抑制剂,用于器官移植和骨髓移植中的抑制排斥现象以及自身免疫疾病。厂方质量标准中乙醇及丙二醇的含量采用石英毛细管柱测定,此种色谱柱在国内使用不普及,我们经多次试验,摸索出一较好的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]条件,适用于国内检测,即以GDX-101为固定相,柱长为2 m,采用氢离子火焰检测器,进样口温度为210 ℃,检测器为280 ℃,柱温采用程序升温,氮气为载气,以二甲基亚砜为溶剂,以正丙醇为内标,可同时分离测定环孢素A中乙醇及丙二醇的含量,改进后的方法,乙醇与正丙醇的分离度为3.1,丙二醇与正丙醇的分离度为5.0,符合中国药典1995年版中乙醇量度检查的分离度要求[1],操作简便,结果准确可靠。1 仪器与试药 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]:SP-6890 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]柱:玻璃柱,长2 m,固定相为GDX-101。 乙醇、异丙醇、丙二醇均为[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]纯,二甲基亚砜为色谱纯。 样品:环孢素A胶囊(山地明),由诺华公司提供,批号为187MFD0797;241MFD0797;166MFD0797;483MFD0797;477MFD0797。 标准贮备液及内标贮备液:精密称取[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]级的乙醇及丙二醇2.50及1.25 g分置50 mL容量瓶中,加二甲基亚砜至刻度,摇匀,作为标准贮备液;精密量取正丙醇5.0 mL置50 mL量瓶中,加二甲基亚砜至刻度,摇匀,作为内标贮备液。2 试验方法与结果2.1 色谱条件 采用GDX-101为固定相,柱长为2 m,氮气为载气,采用氢离子火焰检测器,进样口温度为210 ℃,检测器为280 ℃,柱温采用程序升温,即初始为165 ℃,保持12 min,以40 ℃。min-1升至280 ℃,并保持20 min,检测器温度为280 ℃,进样量为2 μL。2.2 分离度试验 称取乙醇、丙二醇及正丙醇各50 mg置同一50 mL量瓶中,加二甲基亚砜至刻度,摇匀,进样2 μL,按上述色谱条件试验,记录色谱图,见图1-A,乙醇、丙二醇及正丙醇的保留时间分别为1.15,2.22,7.54 min,计算乙醇与正丙醇及丙二醇与正丙醇的分离度,其分离度分别为3.1和5.0。图1 分离度色谱(A)及样品测定(B)色谱图1.乙醇 2.正丙醇 3.丙二醇 4.二甲基亚砜2.3 线性范围及标准曲线 分别精密量取乙醇和丙二醇标准贮备液1.0,1.5,2.0,2.5,3.0 mL,分别置50 mL量瓶中,并分别加入内标贮备液1.0 mL,使乙醇终浓度为1.0,1.5,2.0,2.5,3.0 mg.mL-1,丙二醇的终浓度为0.5,0.75,1.0,1.25,1.5 mg.mL-1,分别进样2 μL,以乙醇及丙二醇的进样量为横坐标,以它们的峰面积与内标峰面积之比为纵坐标,分别进行线性回归,结果线性关系良好,乙醇、丙二醇回归方程分别为:A=8.935×103C+7.858×102 r=0.998 8A=8.086×103C-1.649×102 r=0.999 92.4 精密度试验 用乙醇与丙二醇浓度分别2.0及1.0 mg.mL-1的溶液,重复进样5次,结果乙醇与丙二醇的RSD分别为0.7%和1.0%,精密度良好。2.5 回收率试验 采用加样回收法,取已知乙醇与丙二醇含量的样品2粒,用二甲基亚砜溶解,置50 mL量瓶中,精密加入内标贮备液1.0 mL,并加二甲基亚砜至刻度,摇匀,精密量取此溶液4.0,4.5,5.0,5.5,6.0 mL,分别加入乙醇与丙二醇的浓度分别为2.0 mg.mL-1及1.0 mg.mL-1的标准溶液6.0,5.5,5.0,4.5,4.0 mL,混匀,量取混匀后的溶液2 μL,注入[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],测定这5份溶液的乙醇和丙二醇含量,计算回收率,乙醇的平均回收率为99.9%(RSD<0.8%,n=5),丙二醇的平均回收率为101.4%(RSD<1.1%,n=5)。2.6 样品的测定 取乙醇和丙二醇标准贮备液2.0 mL,内标贮备液1.0 mL,并加二甲基亚砜至刻度,摇匀,作为对照品溶液;取环孢素A胶囊2粒,置50 mL量瓶中,用二甲基亚砜溶解,精密加入内标贮备液1.0 mL,并加二甲基亚砜至刻度,摇匀,作为样品溶液;分别量取对照品溶液和样品溶液各2 μL,注入[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],按上述色谱条件测定,以内标法计算含量,即得;见图1-B。2.7 对比试验结果 取环孢素A样品5批,用改进后的方法测定样品中乙醇和丙二醇的含量,与厂方测定数据相比,结果基本吻合,见表1。表1 乙醇和丙二醇对比试验结果(%) 批号 本法结果 厂方测定数据 乙醇 丙二醇 乙醇 丙二醇 187MFD0797 101.0 106.3 100.5 105.0 241MFD0797 99.2 99.2 100.6 100.6 166MFD0797 101.7 102.7 101.3 103.0 483MFD0797 98.8 96.8 99.3 97.2 477MFD0797 99.1 98.1 98.9 97.7 3 讨论3.1 本法与原厂方方法相比,方法更为简便,条件普及,有利于对样品质量的控制。3.2 原厂方标准在测定乙醇含量时,以正丁醇为溶剂,由于正丁醇的保留时间与丙二醇过于接近,分离度达不到要求,本法采用二甲基亚砜为溶剂,不影响样品的溶解,同时使丙二醇与二甲基亚砜的分离度符合定量分析的要求。3.3 曾用固定相为GDX-401的[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]柱进行检测,乙醇与正丙醇得到完全分离,但丙二醇与溶剂峰重叠,分离度达不到要求。3.4 采用程序升温,可使溶剂出峰时间加快,缩短分析时间。王俊秋(北京市药品检验所 北京 100035)庞青云(北京市药品检验所 北京 100035)余立(北京市药品检验所 北京 100035)参考文献1,中国药典.1995.二部:附录44
请大家说说一般两个主色谱峰分离不开来可以采取哪些方法?
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分离异丙醇和水用什么方法合适?GDX-301填充柱可不可以分离?
液相色谱中死时间几种测定方法1:有响应的溶剂出峰时间为死时间。2:进样后的阀切换峰,对应的时间为死时间。3:工作站中输入色谱柱的空余体积或孔隙率,自动计算。(对于C18柱,也可以用硝酸钠或硫脲等在色谱柱上完全不保留组分的出峰时间来测定死时间。)影响分离度的因素有三个因素控制两个色谱峰之间的分离度——容量因子,选择性,柱效容量因子反映样品分子和固定相及流动相之间的作用力,选择性是说明色谱系统区分两个或多个色谱峰的能力,柱效与色谱峰的宽度有关,很明显要达到一定的分离度,宽色谱峰要比窄色谱峰需要更大的分离度选择性。理论塔板数越高柱效越高,柱效的高低受柱内效应和柱外效应的影响。选择性是固定相区分两个被分离样品组分的能力,用容量因子之比进行计算,它是两个被分离色谱峰顶点距离的量度,如果选择性是Ⅰ,则两个组分完全不能分离。选择性数值越高,分离越好。由于选择性取决于被分离物的物理和化学结构,流动相和固定相,流动相组成,PH,色谱柱温度,流动相添加剂,因此,尽量优化实验条件提高选择性以降低成本。容量因子为物质的特性,当分析条件一定时,容量因子为固定值。溶剂的洗脱强度与其极性有关:反相色谱:溶剂的极性越强,洗脱强度越弱。正相色谱:溶剂的极性越强,洗脱能力越强。(注意:不可以使用纯水作为正想色谱的流动相)一般样品分析要求:容量因子大于2小于5
样品:胖丁丁,Luna样品性质点评,胖丁丁高大威猛略胖,Luna形象气质佳……(只是为了剧情需要,大家多包涵,我吐口先 :)反相柱分析机理:色谱柱:为一间屋子,有一门可进,一门可出,屋里有大群美女。结果:众美女都喜欢帅哥,不断有人拉Luna的手,并要求合影签名等等,拉胖丁丁的少了些,结果胖丁丁和Luna的距离越来越远,出门的时候,已经分离的很好拉,分离度3.0,柱效15万/m反相柱使用范围:1)、不可用于分离帅得离谱的人,会造成美女互相踩伤践踏拥挤的现象,造成柱堵塞,柱压升高;心脏不好的美女过于激动会造成休克,严重者甚至兴奋而死,造成柱子过早老化,降低柱效。另外分离此种物质会造成强吸附现象,出峰时间太久甚至不出峰,2)、不可用于分离过于猥琐丑陋可怕的人,结果会造成美女流失,柱效下降,出峰时间太快,影响分离效果,不过有方法可以恢复柱效,就说此地正莱尔斯丹的鞋正挥泪大甩卖,美女将迅速赶回,柱效即可恢复!注:此恢复方法并不适用于分离杀人犯强*犯!2、正相柱分离原理色谱柱:为一间屋子,有一门可进,一门可出,屋里有大群男子。结果:Luna被率先赶出,胖丁丁被同胞悻悻相惜,留下来吃完饭,吃完后大家含泪送别,分离度2.8,柱效13万/m正相柱分离注意事项:并不适用于分离BT男3、体积排租色谱分离原理色谱柱:钻溶洞结果:溶洞里洞有大有小,非常好玩,本以为Luna个头小灵活会早点爬出来,其实是体积庞大的胖丁丁先爬出来拉:)分离度2.5,柱效12[f
色谱分离原理比喻样品:胖丁丁,Luna样品性质点评,胖丁丁高大威猛略胖,Luna形象[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]佳……(只是为了剧情需要,大家多包涵,我吐口先 :)1、反相柱分析机理:色谱柱:为一间屋子,有一门可进,一门可出,屋里有大群美女。结果:众美女都喜欢帅哥,不断有人拉Luna的手,并要求合影签名等等,拉胖丁丁的少了些,结果胖丁丁和Luna的距离越来越远,出门的时候,已经分离的很好拉,分离度3.0,柱效15万/m反相柱使用范围:1)、不可用于分离帅得离谱的人,会造成美女互相踩伤践踏拥挤的现象,造成柱堵塞,柱压升高;心脏不好的美女过于激动会造成休克,严重者甚至兴奋而死,造成柱子过早老化,降低柱效。另外分离此种物质会造成强吸附现象,出峰时间太久甚至不出峰,2)、不可用于分离过于猥琐丑陋可怕的人,结果会造成美女流失,柱效下降,出峰时间太快,影响分离效果,不过有方法可以恢复柱效,就说此地正莱尔斯丹的鞋正挥泪大甩卖,美女将迅速赶回,柱效即可恢复!注:此恢复方法并不适用于分离杀人犯强*犯!2、正相柱分离原理色谱柱:为一间屋子,有一门可进,一门可出,屋里有大群男子。结果:Luna被率先赶出,胖丁丁被同胞悻悻相惜,留下来吃完饭,吃完后大家含泪送别,分离度2.8,柱效13万/m正相柱分离注意事项:并不适用于分离BT男3、体积排租色谱分离原理色谱柱:钻溶洞结果:溶洞里洞有大有小,非常好玩,本以为Luna个头小灵活会早点爬出来,其实是体积庞大的胖丁丁先爬出来拉:)分离度2.5,柱效12万/m原因:两人一钻溶洞便发现仿佛回到了童年,逮着洞就想钻,不过胖丁丁突然发现自己已不是3岁时的胖胖拉,要是卡住不崴了嘛:)于是只好作罢,沿大路走出来了,扼腕叹息“时光蹉跎,青春少年已不复4、离子对色谱色谱柱:为一间屋子,有一门可进,一门可出,屋里有大群美女。胖丁痛苦回忆:众美女都喜欢帅哥,不断有人拉Luna的手,并要求合影签名等等,拉胖丁丁的少了些,结果胖丁丁和Luna的距离越来越远,出门的时候,已经分离的很好拉,分离度3.0,柱效15万/m胖丁对策:往事不堪回首,所以第二天再过这间屋子的时候,胖丁想到了他的必杀技——小胖。结果,胖丁抱着小胖和Luna一起穿过,众美女发现居然还有如此帅的小男生,纷纷过来掐掐小脸蛋,小胖搭讪美女的工夫也不含糊,“美女,敢吃青椒吗?”。老胖也不手软,为小胖报仇,把众美女脸蛋一一掐个遍 ,正当老胖还色迷迷的看着众美女的时候,Luna已经顺利离开了美女屋,最后小胖发话拉“爸爸,我饿!”老胖才恋恋不舍的抱起小胖,发话“最后再重掐一遍!……”Luna在门口,顿倒……拍摄花絮:1)观众问:美女为什么喜欢小帅哥不喜欢Luna?女人总是有母爱的,这是与生俱来的本能。所以此处美女年龄要大点:)2)排完这段之后,导演“卡”了N次,因为小胖被掐后没有表现出天真浪漫可爱的样子,反而哭了N次,最终排得小胖又累又饿又疼才终被导演放行!3)该CASE结束时,镜头正面是胖斑得意而归的表情,远端发现众美女正在揉脸,忿忿曰“死胖子,手够狠啊!……5555555!”影片悬念:胖斑得意是因为给小胖报仇呢?还是因为别的什么呢?……

http://ng1.17img.cn/bbsfiles/images/2012/04/201204151854_361520_1638724_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/04/201204160801_361554_1638724_3.jpg你认为正、反相色谱分离的原理是什么,吸附、分配还是兼而有之,你是否根据分离原理设计开发过液相色谱分析方法,很多培训资料,教科书中的方法开发介绍时几乎没有从分离机制方面入手的,分离机制在方法开发中是否无用处呢?







 我要推广仪器
我要推广仪器
 下载APP
下载APP