安捷伦液相色谱,多通道的梯度阀(MCGV)有一醋酸水的控制阀关不死。用热水冲了8个小时都不见效果。还有什么好方法。
想做三个流动相的梯度,其中两个变化趋势一致,可以将它们混合,之后当成二元来做吗,结果一致吗?
二元高压梯度(双泵+在线混合器)与四元低压梯度(单泵+低压梯度阀+脱气机+混合器),对于上面两个配置,现在哪个实用性更好?哪个洗脱效果更好?一般首选哪个配置? 顺便弱弱的问一下,如果我选二元高压梯度,对于像甲醇:水:冰醋酸(48:52:1)这种三种溶剂甚至四种的流动相该怎么去弄呢?它总共才二个溶剂通道吧。。。。
請問等度和梯度的分別是什麼?它們的用途有什麼不同?謝謝賜教.
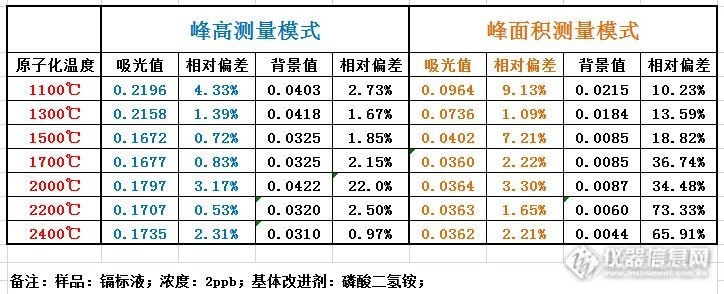
看到本版冰山版友关于石墨炉测镉的温度梯度试验http://bbs.instrument.com.cn/shtml/20140117/5160468/,感到很有意思;于是我利用今天下午半天的时间,用石墨炉对镉的单标液(2ppb)进行了一个原子化温度梯度的试验。一方面重新演绎一遍冰山版友的试验,另一方面也想通过我的试验,能给大家提供一个有趣味的话题。仪器:ZA3000型背景扣除方式:塞曼升温程序:干燥:80°~140° 30秒;灰化:400°~400° 15秒;原子化:1100°~2400°之间改变 5秒;清除:2000°~2500°。石墨管:C型高阻石墨管样品:Cd标液,浓度2PPb,进样量 20微升,每种测量模式重复三次测量。基体改进剂:磷酸二氢铵(2%),进样量5微升。http://ng1.17img.cn/bbsfiles/images/2014/01/201401231636_488710_1602290_3.jpg分析提示:(1)采用峰高测量模式,1500°和1700°是最佳结果,相对标准偏差最小;所以这也就是文献推荐的温度的出处。(2)峰高模式下,不同的原子化温度所得到的吸光值并不是一味地下滑,而是呈现U形。(3)采用峰面积模式吸光值也不是一味地下滑,在1700度~2400度之间基本保持不变,呈现L形。讨论问题:为何采用不同的测量模式所得到的吸光值的变化趋势不一致?
大家做液质分析在选择液相条件时一般是用恒流的多还是梯度洗脱用的多呢?恒流与梯度洗脱两者是如何选择的?什么情况下考虑用梯度洗脱呢?
柱温在液相色谱梯度洗脱过程中扮演重要的角色,提高柱温可以缩短保留时间;柱温还可以影响选择性;温度的不平衡会导致峰扭曲变形等等,那么到底能影响到个什么程度?各位有经验的老师具体讲解一下吧?http://simg.instrument.com.cn/bbs/images/brow/emyc1010.gif
为什么跑梯度的时候总是出现这种基线不稳的情况。[img]https://ng1.17img.cn/bbsfiles/images/2019/05/201905051222265990_538_3501642_3.png[/img]
HPLC方法: 乙腈-水系统; 2.1mm*100mm的小直径的柱子; 流速=0.2mL/min; 梯度: 0-5min: 10%-20% 乙腈; 5-10min: 20%-90%乙腈结果发现化合物保留时间变化很大,而且系统压力不稳定. 工程师来校准后,高流速(1mL/min) 压力稳定,但是低流速压力和保留时间还是很差.难道是5-10min梯度变化太大,低流速时泵的梯度精度不够?!请有经验的朋友给解释一下啊!
上一篇帖子,图乱了,重发一下。看了一篇论文,想复刻它的冲序列。写了一个和论文中一样的脉冲序列,但谱图无法调平,按照它里面给出的梯度场实验,结果是所有质子全部去相。根据它的描述,改进后的脉冲序列,谱图可以调平,但其梯度场依然使全部质子去相。问,有什么解决办法或者合适的梯度场嘛?前面是论文中的脉冲序列和谱图[img]https://ng1.17img.cn/bbsfiles/images/2023/08/202308071200467427_7789_4047002_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2023/08/202308071200471210_8607_4047002_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2023/08/202308071200471367_8815_4047002_3.png[/img]
听说HILIC平衡时间长,又没有实例,走梯度比普通柱子大概要多平衡多久呢?
实验用到梯度洗脱,A:40mM磷酸盐缓冲溶液 B:乙腈:甲醇:水(45:45:10)时间(min)B(%)001.9018.15718.610022.310023.20260请教大家:梯度之前的平衡,要用哪一相平衡柱子?还是要运行几个空白梯度进行平衡?我用的是A相,直至压力和基线稳了才进行梯度分析,这样对吗?关于梯度洗脱柱平衡的注意事项请大家多指教一下。
A-甲醇;B-0.1%甲酸梯度:0-5min,10%A5-10min,10-15%A10-18min,15-20%A18-23min,20-30%A23-28min,30-35%A28-35min,35-40%A35-45min,40-50%A45-50min,50-10%A50-55min,10%A56min,停止我的样品跑出来的图中间有一段基线是弧形的,就是一堆峰都没有落回到0,但是因为里面没有我要的峰我就没管,后面我要的峰还可以。最近要做方法验证,做定量限检出限的时候发现,从8min开始,柱子的压力一直在上升,到了26min开始平稳,到了48min开始下降,我的基线也是这样的波动趋势。问题是我的标品在20多分钟出峰,因为基线在这一段是往上飘的,所以带着我的标品一起上去了,最后出来的谱图很难看,(之前的应该是样品中含量大,所以把波动给压平了?)而且还识别不出来。想问问大家这该怎么办啊?
大家好,我有一套岛津HPLC,工作站是LC-Solution,2个LC-10AD 泵组成高压梯度,但是是当我检查梯度准确性是,发现线性梯度的曲线是弯曲的,而我的梯度曲线值“curve” 是默认的“0”,2个泵流速是准确的,请问是不是梯度曲线的问题啊?改设置为多少呢?
小弟作验证时梯度变化比较快,基线不稳(没有办法改进),噪音很大,对LOD,LOQ影响很大,请教有什么办法,是不是扣除空白后再算基线?
各位老师好,我们实验室一台LC-10ADVP的液相异常,仪器是2个高压泵,紫外检测器,混合器,机器故障具体表现是需要很长的时间才能基线平稳下来(0.5-2个小时), 基线平衡了后分析样品,开始基线都正常,但是当梯度发现变化的时候,基线就不好了, 会呈现有规律的出峰, 而且观察流动相有规律的气泡,需要在流动相梯度没有变化的时候再平衡蛮久的时间才好。 如果不用梯度,只用单泵工作, 不管是A泵还是B泵都显示是正常的没有类似的。 开始拿去维修,怀疑是单向阀不干净这类的问题,也或怀疑是单向阀宝石球的问题(因为基线不平衡的问题针对乙腈非常的明显, 甲醇则情况好很多,但是甲醇也有基线重新不平衡的问题),清洗了泵单向阀更换了一个垫片后问题并没有解决(维修方只是清洗更换单向阀垫片后走了空白溶剂,平衡后略微改变梯度是波动小,)但是我们拿到机器走样的时候还是和之前差不多 当梯度有变化的时候就很明显的基线波动, 有流动相有气泡,我想请教下老师碰到这种情况怎么排查和缩小故障的范围, 我们自己是担心是不是混合器的问题,如果是从那个地方排查呢?
看了一篇论文,想复刻它的冲序列。写了一个和论文中一样的脉冲序列,但谱图无法调平,按照它里面给出的梯度场实验,结果是所有质子全部去相。根据它的描述,改进后的脉冲序列,谱图可以调平,但其梯度场依然使全部质子去相。问,有什么解决办法或者合适的梯度场嘛?前面是论文中的脉冲序列和谱图后面是修改后的脉冲序列和谱图[img]https://ng1.17img.cn/bbsfiles/images/2023/08/202308071148572663_9447_4047002_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2023/08/202308071148574883_7289_4047002_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2023/08/202308071148574402_4424_4047002_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2023/08/202308071148577090_7216_4047002_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2023/08/202308071148577793_9866_4047002_3.png[/img]
因我的色谱峰有一杂峰干扰药峰,我想用梯度洗脱将其分离出去,但目前我的流动相含有SDS(十六烷基硫酸钠),因加有离子对试剂的不易平衡和稳定,我想问问梯度洗脱会不会影响稳定性。比如说会不会大大影响药峰的保留时间、峰面积,等等。请各位高手指教。。。
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=111363]液相色谱梯度洗脱中柱温的影响[/url][em0815]
[align=center]开发了一个梯度方法,当我在我的系统上运行它重复性很好,但我不能在另一个系统上得到相同的结果?[/align]当梯度法从一个高效液相色谱系统转移到另一个系统时,偶尔会出现一些困难。除非系统完全相同,否则用户通常会在保留时间上发生一些变化。大多数情况下,留存率的变化不会影响到解决方法的效果,人们通常可以忽略这些差异。另一方面,通过对潜在原因的正确理解,我们可以调整梯度,以从不同的HPLC系统获得相同的性能。梯度驻留体积:它是梯度混合点和柱顶之间的体积。在你通过注入样品开始分析后,梯度将不会到达柱的顶部,直到梯度停留体积被清除。这意味着你的样本最初要经历一段等稳迁移的时期,直到梯度赶上。由于不同系统的梯度停留体积不同,这种等稳偏移时间也会不同,可能导致滞留时间的差异,甚至影响分辨率。另一种可能是梯度本身。系统与系统之间可能存在组成差异。对于今天生产的大多数HPLC系统,这应该只是次要的问题。但是,一般来说,任何梯度系统都能在成分的中间范围,即50% a和50% B的混合物中,提供具有最高准确度的成分。当非常不成比例的a和B混合时,例如5% a或95% a,准确性就会受到影响。[align=center]哪些可能导致我的方法传输问题?[/align]最简单的事情是比较你的梯度两个系统。为了做到这一点,你断开色谱柱,并在梯度的b溶剂中添加紫外线吸收剂。如果你使用的是反相体系,你可以在b溶剂中加入10mg /L的对羟基苯甲酸丙酯。然后在两个系统上运行渐变并记录基线。然后将这两个情节相互比较。你需要找到梯度开始的点,你还需要测量梯度剖面。如果你的梯度是线性的,那么你只需要检查梯度的斜率。很有可能你会发现两种仪器之间的梯度开始是不同的,而轮廓是非常相似的,只是被一些时间抵消了。在这种情况下,你有一个不同的梯度停留体积。[align=center]是否有一种简单的方法来补偿驻留体积的差异?[/align]有一个解决方案大多数时候都是有效的。如果梯度停留体积在你的方法转移到的系统上较小,你可以通过在梯度开始时设计一个补偿体积差异的等稳部分来补偿停留体积的不足。剩下的梯度保持不变。另一方面,如果第二个系统的梯度停留体积比第一个系统的梯度停留体积大,则情况更困难。原则上,您可以启动梯度,然后在延迟一段时间后注入样品,这段时间可以解释两种系统之间梯度停留体积的差异。但在自动系统上,这可能是不可能的,因为注入会触发梯度的开始。在这种情况下,您可能需要回到原点并重新开发该方法。[align=center]我能做什么来防止这种情况在未来发生?[/align]您可以通过为最终将使用该方法的HPLC系统开发该方法来避免这种情况。这需要一些远见和一些计划,但通常不是不可能的。您首先需要做的是描述可能用于您的方法的系统。对于每个系统,你需要知道两件基本的事情:梯度驻留体积和合成精度。你可以在一个实验中得到这两个信息:如上所述,你在你的b溶剂中添加一个紫外线吸收剂。然后你在0% B到100% B的增量5%的多个步骤梯度。流量应该是通常使用的流量,所以最有可能你将使用1毫升/分钟的流量。步骤之间的间隔应该是几分钟,或者5分钟。现在在不同的系统上运行这个梯度,而不需要一个柱,并记录检测器的响应。为每一步编程的时间和实际发生的步骤之间的时间延迟给你梯度延迟时间。台阶的高度给了你构图的一个度量。步骤会被抹去一点,这是系统中混合体积的函数。现在您已经确定了系统的特征,您可以围绕系统的特征设计您的方法。正如我之前提到的,最大的问题通常是渐变驻留体积。如果您知道需要转移您的方法的系统比开发您的方法的系统有更大的驻留体积,那么您应该在您的方法开发中自动添加一个等稳步骤,以补偿这种差异。如果目标系统的驻留体积小于开发系统的驻留体积,那么您应该能够通过在转移方法时在方法的开始部分添加一个梯度延迟时间来简单地补偿这一点。如果在渐变过程中存在组成差异,你也可以通过调整渐变轮廓来弥补这些差异,但我从来没有遇到过这种情况。本讨论假设柱与起始流动相处于完全平衡。我偶尔会遇到这样一种情况,在常规分析中,梯度彼此跟随得如此之快,以至于柱在起始流动阶段永远不会恢复平衡。你会发现,如果你的第一个梯度总是给出不同于后续梯度的结果,你就会遇到这种情况。这可能有利于加快方法的速度,但当将该方法转移到具有不同驻留体积的另一个系统时,这可能会造成困难。
液质联用精密度问题在做液质联用定量分析中药中多种成分时,以A:0.3%甲酸水,B:乙腈为流动相,采用梯度洗脱,洗脱程序:0-6min 20%-40% B,6-8min 40%-100% B,8-14 min 100% B,13 min内所定量成分能完全出峰,主要是香豆素和生物碱类,以前的液相条件在质谱上分离挺好,但20 min之后保留时间漂移较大,也曾用等度洗脱试,峰分不开,采用现在的梯度条件,保留时间基本稳定,做好线性关系后,精密度考察出现问题,采用多反应离子监测,每次所测含量呈不规律变化,有时高有时低,进样20多针相同浓度的混标液精密度依然很差,RSD能达到20多,有的成分含量前后能相差1倍,用的是Agilent1200的液相,6410 QQQ-MS色谱柱是2.1×150 mm,1.8u的,流速是0.3 ml/min,混标液是用部分甲醇和部分流动相溶的,仪器也是前段刚检过的,都pass了,不知什么原因,是每次离子化程度不一样吗,请各位高手指教。还有做精密度、重现性、回收率时,每次都要做一次线性关系吗
[color=#444444]最近在开发一个方法检测一个复方药,想用梯度液相分离两个原料药。条件如下,流动相A 10mM醋酸胺溶液,流动相B 0.1% 甲酸甲醇溶液。[/color][color=#444444]梯度为0-5分钟 流动相A从90%降为0%,5-6 分钟保持流动相A 0%, 6.1-9分钟回到初始条件平衡。[/color][color=#444444]用柱子为沃特世Sunfire C18, 4.6* 50mm, 3.5um的柱子,柱温为30度。检测波长273nm.[/color][color=#444444]得到的溶剂图如下,曾经怀疑是溶剂不干净,用纯水跑过,得到的色谱图和溶剂一样,也是这么多杂峰。[/color][color=#444444]请教各位大神,有什么原因会造成基线有这么多杂峰呢?我需要怎么改进呢?[/color][color=#444444][img]http://muchongimg.xmcimg.com/oss2/img/2018/0815/w75h8461321_1534343279_714.png[/img][/color][color=#444444][img]http://muchongimg.xmcimg.com/oss2/img/2018/0815/w75h8461321_1534343285_410.png[/img][/color][color=#444444][img]http://muchongimg.xmcimg.com/oss2/img/2018/0815/w75h8461321_1534343291_927.png[/img][/color][color=#444444][img]http://muchongimg.xmcimg.com/oss2/img/2018/0815/w75h8461321_1534343295_390.png[/img][/color][color=#444444][img]http://muchongimg.xmcimg.com/oss2/img/2018/0815/w75h8461321_1534343296_520.png[/img][/color][color=#444444][/color]
用液相色谱测定TBHQ和BHT,方法是采用SN/T 1050-2002采用的是梯度洗脱的方法色谱条件a)色谱柱: C18柱〔250mmX4.6mm) b)柱温: 4 0°Cc)流动相: 1.5%(体积分数)乙酸-甲醇溶液(A),1.5 %(体积分数)乙酸—水溶液(B)d)洗脱梯度: 0min -5min ,流动相A为30%,5min-20min,流动相A从30%线性增至80%,20min -30min,流动相A为80% e)流速: 1 m L / mi n f)检测波长: 2 8 0用得是岛津的液相色谱,工作站是class-vp我是这样设置的 Time ModuleAction Value5.01pumpsB.Conc7020.01pumpsB.Conc2030.01pumpsB.Conc2036.00SPDstop最后一栏的Module是不是SPD不太记得了 因为在休假有点不记得了 [em09511]总之就那一个Module的选项里有stop测定后只有TBHQ出峰 BHT没有出峰我想应该是这个梯度编写有问题但是说明书上米有详细的步骤所以请教各位大大们我这样写有问题哇能给个梯度编程的资料什么的好么我在岛津那一块找了很久么找到[em09512]
分析方法前十分钟设置等度运行。压力稳定,十分钟后运行梯度每四分钟出现一次锯齿状压力波动
大家看我这个梯度走出来的图,为什么15分到22分之间有这么多峰呢,这是纯溶剂的空白图,梯度大概是,A为95%的缓冲盐,B为5的乙腈,0-30分钟,A从95降到60,30到31分钟,A从60升到95,31到40分钟A95走10分钟
各位大虾们,我用的是Agilent1100,做外包服务,化合物纯度在95%以上就可以交差了,以前直接看化合物的性质选择流动相,比如,如果化合物带酸根的我就选择带酸性流动相,后来觉得这样找条件太费时间了,而且有些化合物看不出酸碱性,就试试梯度走,我走的样品是酸性的,就用ACN:0.1%甲酸(梯度如下),后来对照时间,感觉主峰是100%乙腈冲出来的,而且杂质也多,基线也不平,后选择ACN:0.1%甲酸(70:30)重新走样也把主峰给走出来了,不过杂质没先前的多。请问梯度的主要用途以及怎样的设置法?时间 ACN 0.1%甲酸 2min 20 8015min 100 018min 20 80
0即样品顺离心力方向沉降σ〈 S时 V〈 0即样品逆离心力方向上浮σ=S时 V=0即样品停止沉降或上浮,"稳定"在这一位置用这个公式可以很好地解释在速率一区带离心法或等密度离心法中单一样品的沉降(或上浮)行为。二、 转头的选择:1、离心转头分类:转头类别 使用的离心机 发明时间、发明者或推广商固定角式转头 低、高、超速 1943年,(英)Pickels甩平转头 低、高、超速 1951年,(德)Kahler垂直管转头 高、超速 1974-1975年,(美)Dupont公司区带转头 低、高、超速 1964-1965年,(英)Anderson、(英)MSE公司近垂直管转头 超速 1989年,(日)Hitachi Koki、(美)Beckman公司连续离心转头 低、高、超速 1965年,(英)MSE公司其他特种转头:分析转头,土壤脱水转头,细胞浮选转头,管式转头,血球比测定转头,细胞清洗转头等等。2、 各种转头用于密度梯度离心的比较:(I) 各种转头用于速率一区带(R-z)离心的优缺点分析:固定角式转头:壁部放应影响很大,用于R-z离心回、收率低,纯度也受影响一般知用于差分离心和等密度离心,离心时间向较短。是各其离心机的最高速转头。甩平转头:细长离心管用于R-z离心可以获得较高纯度和高分辨率,且容易控制离心时间,壁部入在很少。25000rpm~30000rpm的甩平转头最适用于亚细胞器的离心分离,而40000~42000rpm的甩平转头适合用于核酸、蛋白、病毒草类物质内流的分离。离心时间较长。垂直管转头:沉降距离最短,因而离心分离时间也是短。最大半径前几乎没有壁部放位,最大本径后右一定壁部效位,垂直剖面积较大,因而离心后纯样品区带的容量也较大。但在有沉殿的密度梯度离心中,沉淀和浮动区带方向转换之间存在干扰,可能影响纯样品区带的纯度。大部份R-z离心没有沉殿,垂直管转头很适合做R-z离心。近垂直管转头:管轴线与旋转主轴之间倾角7度~9度(角式转头20度~45度)沉降距离比垂直转头稍大,离心时间比角式,甩平转头都要短。由于右了倾角,沉殿可沿管壁滑向低部,因此基本上清除了沉殿与浮动区带转换之间的干扰,适合做R-z离心,特别适合做生物大分子(如质粒DNA)的自形成梯度等密度离心。区带转头:没有壁部入应特别适合做大容量的病毒,亚细胞器,生物大分子的R-z离心,可用于研究,中试和少批量生产。分离纯度高,量大,但操作要求高,转头及整体价格昂贵。连续流离心转头:工作原理的区带转头相似,可连续工作,分离量大,分离统纯度高,可用于各种生物体的差分,R-z等密度离心。近年来高速连续流转头常用于大量发酵液(大肠样菌、酝母菌)菌体的沉殿。(II) 各种转头用于等密度离心内优缺点分析:固定角式转头:主要用于差分离心的角式转头,在速离心机上可以很好地用于等密度离心,尤其是DNA平衡等密度离心,自形成梯度,用快速密封管或厚壁管,常用的单管的容量为10~15ml,常用转速40000~60000rpm,离心时间较短,分离纯度也较高。甩平转头:用作等密度离心时,壁部效应对分离效果影响较少,梯度变换在甩平时自然过渡因而很适合做等密度离心实验,优点是回收率高,分辨能力强。缺点是沉降距离长,最高转速较低(由于结构层固此类转头最主转速一般在60000rpm以下)因而,离心时间很长,对某些长离心管,10~15ml容量的转头最高转速在40000~41000rpm,用作自形成CSCL梯度的质粒DNA离心往往需要50~70小时。垂直管转头:适合作等密度离心,沉降距离最短,在没有沉殿或沉殿非常坚实的情况下,对于现代可自由选择加速、减速时间的离心机,梯度转换得很好。这类转头转进很高(目前最高转速可达100000rpm,70000xg)离心时间最短,分离纯度高,样品容量较大(垂直剖面积最大)。近垂直转头:九十年代以后开始使用的新型转头,用作生物大分子(DNA,RNA,蛋白质等)的平衡等密度离心最佳,目前这类转头的最高转速已达90000rpm近650000xg),离心时间相比垂直转头稍长。区带转头:非常适合做大容量样品的等密度离心。连续流离心转头:高、低速连续流转头一般不用作密度梯度离心超速连续流转头适合做大容量样品的等密度
请教:走等度时基线很平稳,而走梯度时基线45度角上飘,为什么?流动相是0.1%三氟乙酸水与0.1%三氟乙酸乙腈
化工行业,合成的产品和里面含有的主要杂质如图所示流动相:乙腈:水,pH=7,梯度洗脱有几点请教大家:1.梯度洗脱为什么进一针纯水会出现这么大的基线波动?2.走样子为什么杂质峰会这么多?3.该怎样选择此物分离的流动相条件?需要加缓冲盐、离子对或者调pH吗?主要物质结构http://ng1.17img.cn/bbsfiles/images/2015/09/201509161640_566311_3038012_3.jpg空白梯度http://ng1.17img.cn/bbsfiles/images/2015/09/201509161641_566313_3038012_3.jpg空白梯度放大http://ng1.17img.cn/bbsfiles/images/2015/09/201509161641_566312_3038012_3.jpg梯度18' 0-100%乙腈http://ng1.17img.cn/bbsfiles/images/2015/09/201509161641_566315_3038012_3.jpg上图放大http://ng1.17img.cn/bbsfiles/images/2015/09/201509161641_566314_3038012_3.jpg

自从1977 年推出以来,二氧化硅胶体PercollTM已经成为全世界数以千计的研究人员对密度梯度介质的选择。其近乎完美的物理特征方便它在细胞、细胞器、病毒和其他亚细胞颗粒分离中的使用。Percoll做为第一步在进行更高分辨率分离或核酸抽提前富集细胞是非常有用的。人们会认识到在进行其他的这些方法前使用Percoll 做为第一步可以节省大量的时间和资源。对于生物学颗粒,理想的梯度培养基被描述为具有以下特征: 涵盖了足够的对于所有感兴趣的生物颗粒的恒定密度(图1) 带范围 拥有生理离子强度和pH 在全部梯度中是等渗的 低粘度 无毒性 不会渗透生物膜 无菌且可以重复灭菌 在适度的离心力下将自动形成梯度 和生物材料相容 很容易从被纯化的材料中去除 不影响分析程序 不会猝灭放射性分析 http://www.biomart.cn//upload/userfiles/image/131225996878281.jpg Percoll在现有的介质中是非常特殊的,它符合上述所有的标准,并且提供以下附加的优点: 它能形成连续梯度和不连续的两种梯度。 梯度的稳定性意味着梯度可以预制以提供可重复性的结果。 使用带颜色的Density Marker Beads进行梯度分析十分简单(GE Healthcare提供)。 Percoll 不影响被分离的材料进一步的研究。 数以千计的研究人员的成功已经记录在Percoll Reference List 中。 密度梯度离心原理当颗粒悬浮液被离心时,颗粒的沉降速率和应用的离心力是成比例的。溶液的物理性质也会影响沉降速率。在一个固定的离心力和液体粘度下,沉降速率和颗粒大小以及它自身密度与周围介质密度之间的差别成比例。在一个离心范围中一个球体的沉降方程为:http://www.biomart.cn//upload/userfiles/image/131226013987183.png这里v = 沉降速率d = 颗粒直径(流体力学等效球体) pp= 颗粒密度p1 = 液体密度 h= 介质粘度g = 离心力从这个方程中,可以观察到下列关系: 颗粒沉降速率和它的大小成比例。 沉降速率和它自身密度与周围介质密度之间的差别成比例。 当颗粒密度等于周围培养基密度时,沉降速率为0。 沉降速率随着介质粘度的增加而降低。 沉降速率随着离心力的增加而增加。 通过密度分离(等密度离心法)在这个技术中,梯度介质的密度范围包含了样品颗粒的所有密度。每种颗粒将沉降到梯度中的平衡位置,在这个位置梯度密度等于颗粒密度(等密度位置)。因此,在此类分离中,颗粒基于不同的密度而被单独分离,与颗粒大小无关。 http://www.biomart.cn//upload/userfiles/image/131226021809484.png图1显示两种类型的梯度分离(见下面的速率区带离心法)。当使用Percoll时,普遍是等密度分离颗粒而不是根据颗粒的大小差别(仅见31 页的图19,两种技术都使用)。注释: 当考虑生物学颗粒时,切记介质的渗透压能够明显地改变膜结合颗粒的大小和表观浮力密度。一个高的渗透压能够导致膜结合颗粒收缩而培养基低的渗透压将导致结合颗粒的膨胀。 http://www.biomart.cn//upload/userfiles/image/131226032040485.png图2 显示在生理条件下( 280 到320mOsm/kg H2O) 使用Percoll梯度离心的颗粒比用蔗糖或甲泛葡胺离心的颗粒有低得多的表观浮力密度 通过大小分离(速率区带离心法)在这类技术中,颗粒之间的大小差别与颗粒的密度一起影响分离。正如上述的方程所示,大颗在整个梯度中比小颗粒移动更快,因此选择密度范围以便在整个分离期间的所有的位置上的颗粒密度大于介质密度(图1)。被分离的区带到达管底部(或者它们的平衡位置) 之前运行被终止。







 我要推广仪器
我要推广仪器
 下载APP
下载APP